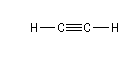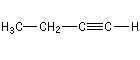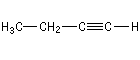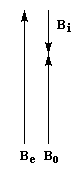Cours de chimie organique - G. Dupuis - Lycée Faidherbe de Lille
Les alcynes
Généralités
Définitions
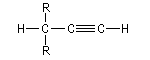
Les alcynes sont les hydrocarbures linéaires de formule CnH2n-2. Le premier terme est l'éthyne ou acétylène. Cet hydrocarbure a été isolé par
préparé par M. Berthelot en 1860. Sa synthèse à partir du carbure de calcium est due au chimiste français H. Moissan. Le carbure de calcium est obtenu par réduction de l'oxyde de calcium au four électrique. Cette réaction consomme une quantité importante d'énergie.
![]()
De nos jours, la préparation industrielle de l'éthyne est effectuée par déshydrogénation thermique du méthane ou de l'éthane à haute température.
![]()
![]()
L'alcyne le plus important dans l'industrie
|
L'éthyne est l'alcyne le plus important sur le plan industriel. Le composé se décompose sous pression.Il est conservé dissous dans l'acétone. Zoom : |
Molécules comportant une triple liaison carbone-carbone
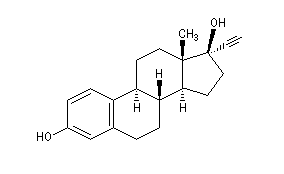 L'éthynyloestradiol est largement utilisé comme contraceptif oral.
L'éthynyloestradiol est largement utilisé comme contraceptif oral.
Zoom : |
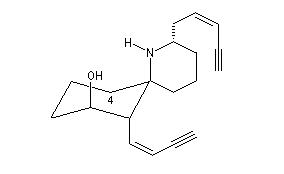 L'histrionicotoxine est un poison produit par certaines grenouilles venimeuses [26].
L'histrionicotoxine est un poison produit par certaines grenouilles venimeuses [26].
Zoom : |
| Le cyclononyne est le premier cycloalcyne non substitué stable à la température ordinaire. Zoom : |
Les cycloalcynes constituent un point de départ intéressant dans la synthèse des fullèrenes comme le buckminsterfullèrene. Du fait de la forte tension de cycle dans le réactif de départ, la transformation suivante est thermodynamiquement favorisée [40].
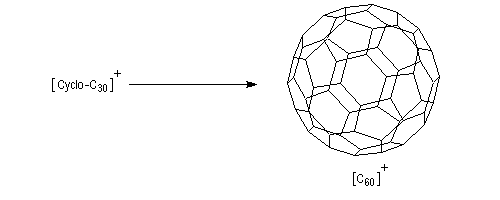
Le benzyne est un cycloalcyne particulièrement instable.
|
|
|
|
|
|
Ethyne |
Propyne |
But-1-yne |
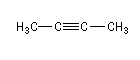
But-2-yne |
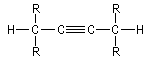
Les composés comportant plusieurs triples liaisons sont appelés diynes, triynes etc.
Structure et propriétés physiques
|
Composé |
Ethane |
Ethène |
Ethyne |
|
d (CC) (nm) |
0,154 |
0,134 |
0,120 |
Propriétés physiques
Les interactions de Van der Waals sont plus importantes que chez les alcènes et les alcanes correspondants car les molécules sont plus compactes. L'éthyne, le propyne, le but-1-yne sont des gaz dans les conditions normales de température et de pression. Les autres sont liquides ou solides.
|
Composé |
Ethane |
Ethène |
Ethyne |
|
T (° C) |
-88,6 |
-103,7 |
-84,0 |
Spectroscopie
Spectroscopie infrarouge
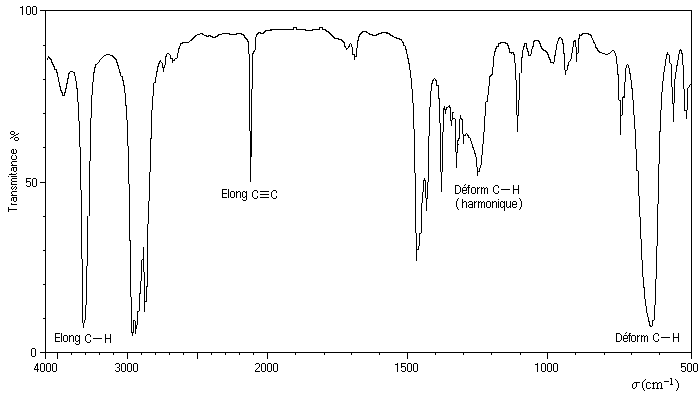
Le spectre ci-dessous est celui de l'hex-1-yne.
|
Vibration |
Elongation C-C |
Elongation C-H |
Déformation C-H |
|
s (cm-1) |
alcynes internes |
3330-3267 |
700-610 |
|
Commentaire |
plus forte dans le deuxième cas |
bande propre aux alcynes terminaux, étroite et intense |
large et intense (harmonique entre 1370 et 1220) |
remarque : la conjugaison avec un groupe carbonyle polarise la triple liaison CC et accroît nettement l'intensité de l'absorption.
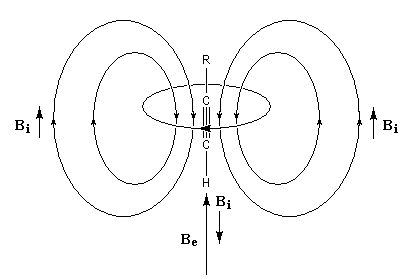
Spectroscopie de RMN Un proton nu, c'est à dire dépourvu d'environnement chimique résonne pour un champ appliqué que nous noterons B0. Bien qu'un atome de carbone digonal soit plus électronégatif qu'un atome trigonal, l'atome d'hydrogène d'un alcyne terminal a un déplacement chimique plus faible qu'un atome d'hydrogène d'alcène. Cela provient d'un effet d'anisotropie du champ magnétique.
Le spectre ci-dessous est celui du 3,3-diméthylbut-1-yne.

|
|
|
|
Composé |
|
|
|
J (Hz) |
(4J) 2-3 |
(5J) 1-3 |
Le spectre ci-dessous est celui du but-1-yn-4-ol.
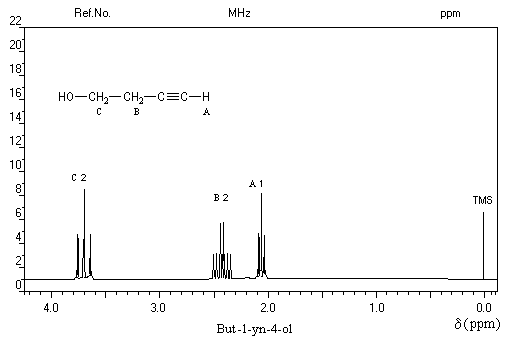
Le massif B centré sur d = 2,4 ppm est un doublet de triplets. Le triplet est dû au couplage avec les protons du méthylène C. Le dédoublement est dû au couplage à longue distance avec le proton A (le proton du groupe OH n'apparaît pas sur ce spectre).
Propriétés des alcynes terminaux
|
Composé |
méthane |
éthène |
éthyne |
|
pKa (couple) |
60 |
45 |
26 |
Les ions alcynure sont facilement préparés par réaction entre un alcyne terminal (souvent appelé improprement, pour des raisons historiques, alcyne vrai) et une base comme l'ion amidure dans l'ammoniac liquide. L'acétylène ou éthyne (pKa = 25) est un acide plus fort que l'ammoniac (pKa = 33). Ainsi l'ion éthynyle peut être préparé en utilisant comme base l'ion amidure dans l'ammoniac liquide.
![]()
Les ions alcynure sont d'excellents nucléophiles. Leur alkylation est une méthode d'élongation de chaîne carbonée.
Les alcynures de Cu (I) et de Ag (I) sont des composés peu solubles dans l'eau. Ils sont instables et dangereusement explosifs à l'état sec.
Synthèse des organomagnésiens alcyniques
Les alcynes étant beaucoup plus acides que les alcanes, l'atome d'hydrogène d'un alcyne terminal est arraché par l'organométallique. On a ainsi une voie d'accès aux organomagnésiens alcyniques qu'on ne peut pas préparer par union directe entre un halogénure et le magnésium.
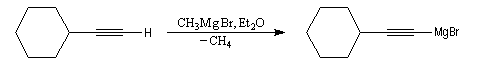 .
.
Les magnésiens dérivant des alcynes sont des réactifs utiles qui permettent l'allongement des chaînes carbonées tout en préservant la possibilité de réactions d'addition sur la liaison triple.
Alkylation des ions alcynure
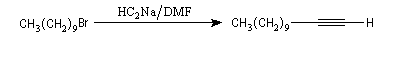
Avec des halogénures de classe plus élevée, l'élimination E2 prédomine ou devient exclusive.
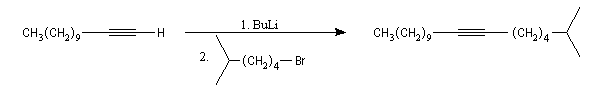
Alkylation des dianions alcynure
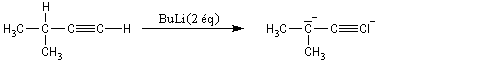
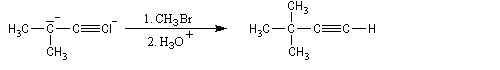
Réduction des alcynes
Addition du dihydrogène
L'hydrogénation en présence de nickel de Raney conduit à l'alcane par l'intermédiaire d'un alcène. Les valeurs différentes des enthalpies d'hydrogénation des alcynes terminaux et substitués isomères, montrent que la substitution par un groupe alkyle stabilise la triple liaison. Cet effet est à rapprocher de celui observé sur la double liaison des alcènes.
|
Composé |
But-1-yne |
But-2-yne |
|
DrHo (kJ.mol-1) |
-292 |
-275 |
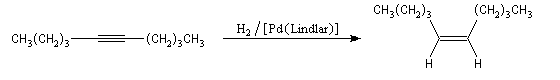
Réduction par un métal
Il est possible d'obtenir un composé éthylénique de stéréochimie E en effectuant la réduction de la triple liaison par un métal alcalin (Li, Na, K).
Cette réduction est stéréosélective de stéréochimie anti. On obtient l'alcène de configuration E.
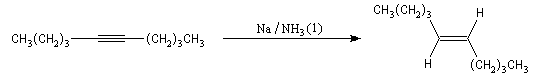
Le mécanisme fait intervenir les étapes suivantes :
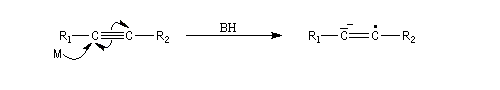
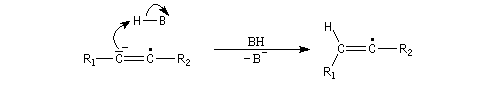
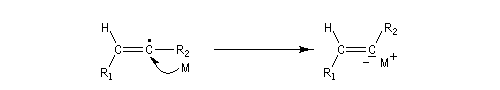
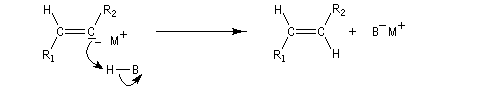
Réactions de couplage
Couplage de Glaser
Cette réaction mise au point par le chimiste allemand Glaser en 1869 permet la préparation des diynes symétriques. Elle consiste à faire réagir un alcyne terminal en présence d'un halogénure de cuivre (I) et de dioxygène. Le cuivre (I) est utilisé en quantité catalytique. Il est oxydé au fur et à mesure de la réaction par le dioxygène.
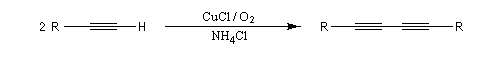
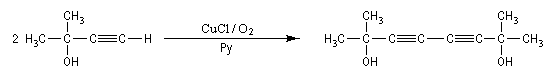
Couplage de Eglinton
Comme la réaction de Glaser, il s'agit d'une synthèse de diynes symétrique à partir d'un alcyne terminal. Cette fois, on utilise une quantité stœchiométrique d'acétate de cuivre (II). La réaction est particulièrement adaptée à la synthèse des molécules cycliques.
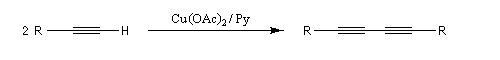
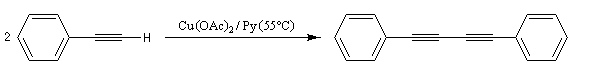
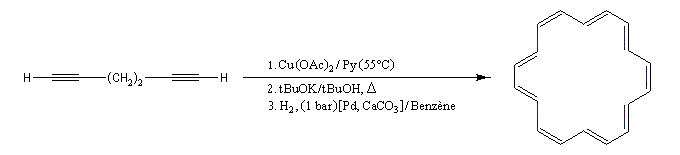
Couplage de Cadiot-Chodkiewicz
Cette réaction, mise au point par les chimistes français P. Cadiot et W. Chodkiewicz (ESPCI et Université de Paris) en 1955, permet la synthèse des diynes dissymétriques. On utilise un halogénure de cuivre (I) comme catalyseur en présence d'une amine.
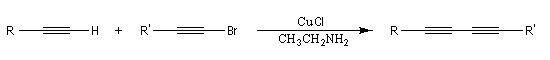
Couplage de Sonogashira
Il s'agit d'une réaction de couplage entre un alcyne terminal et un dérivé halogéné vinylique ou arylique ou un substrat équivalent (triflate). Le catalyseur utilisé est un complexe du palladium (0) ou un précurseur de ce catalyseur sous la forme d'un sel de Pd (II) soluble dans le milieu et réduit en Pd (0) in situ. L'iodure ou le bromure de cuivre (I) sont utilisés comme cocatalyseur. Une amine tertiaire sert de base.
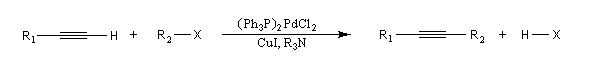
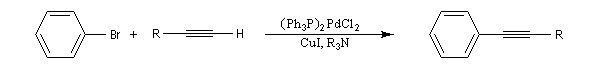
Le mécanisme rappelle celui de la réaction de Heck.
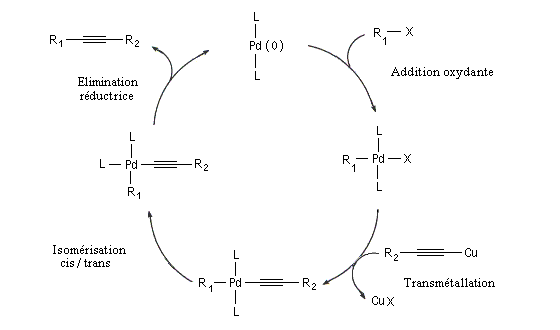
Si l'on utilise un halogénure vinylique de configuration déterminée, la stéréochimie est conservée.
Les réactions ci-dessous constituent les première étapes de la synthèse du galbanolène ou (3E, 5Z)-1,3,5-undécatriène utilisé en parfumerie.
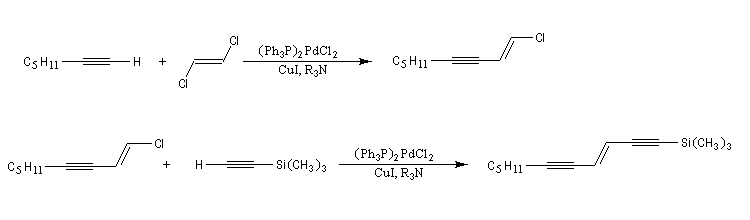
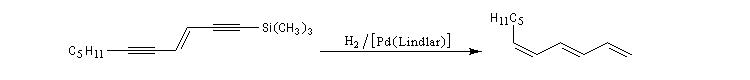
Oxydation avec coupure de la chaîne carbonée
Ozonolyse Additions électrophiles Addition des dihalogènes Addition ionique des hydracides halogénés Le mécanisme fait intervenir un carbocation vinylique. Une des applications de cette réaction est la synthèse du 1,1-dichloréthane, un produit de base de l'industrie des matières plastiques. Addition d'eau Les alcynes disubstitués fournissent des cétones. Remarque : lorsqu'on réalise cette réaction en utilisant de l'éthyne préparé à partir de carbure de calcium, il ne faut pas oublier d'en effectuer la purification en intercalant un récipient contenant une solution de sulfate de cuivre (II) entre le générateur et le réacteur. Le carbure de calcium ordinaire est impur et par action de l'eau il se dégage du sulfure d'hydrogène qui empoisonne le catalyseur (l'affinité des sels de mercure pour les composés soufrés et phosphorés est particulièrement élevée).
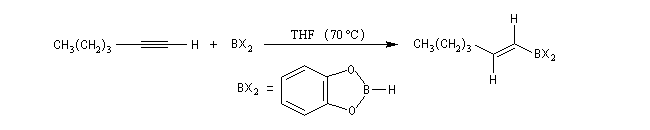
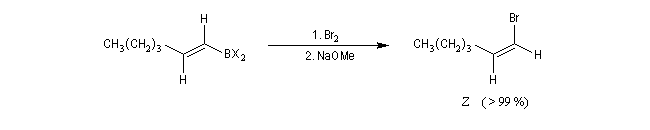
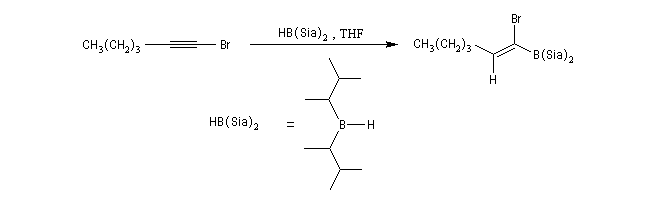
Le mécanisme de ces réactions fait intervenir la formation d'un énol. L'équilibre de tautomérie est en faveur du composé carbonylé plus stable que l'énol. Dans la formation de l'énol, la règle de règle de Markovnikov est suivie. Il se forme de façon intermédiaire le carbocation le plus stable, c'est à dire le plus substitué. Préparation des alcénylboranes Les 1-alcénylorganoboranes peuvent être obtenus avec une excellente stéréosélectivité. La stéréochimie dépend du réactif utilisé. Le 1,3,2-dioxaborole (catécholborane noté BX2 ci-dessous) est préparé en faisant réagir le catéchol (1,2-dihydroxybenzène) avec un équivalent de borane. Avec ce réactif, l'alcénylborane obtenu possède une configuration E. Propriétés des alcénylboranes Ces composés sont aisèment convertis en acides boroniques par hydrolyse. Ces derniers sont facilement convertis en halogénures vinyliques. La bromation des alcénylboranes est stéréospécifique de stéréochimie trans comme pour un composé éthylénique ordinaire. Par addition de méthanolate dans le méthanol, un complexe avec le bore accepteur est formé. L'élimination b stéréospécifique anti qui suit, conduit à un halogénure vinylique de stéréochimie Z. Ces composés sont des intermédiaires de synthèse précieux. Ils sont notamment utilisés dans la réaction de couplage de Suzuki.
Réactifs organométalliques Carbocupration Ces composés sont aussi très utiles pour la préparation d'halogénures vinyliques de stéréochimie bien déterminée.
Hydrozirconation Addition du bromure d'hydrogène Isomérisation des alcynes disubstitués Passage aux allènes Isomérisation Polymérisation Trimérisation Tétramérisation Synthèse du polyacétylène A 150 °C, le polymère cis s'isomérise en polymère trans. Une méthode de synthèse plus récente, consiste à effectuer la métathèse du cyclooctatétraène. Le polyacétylène est un exemple de polymère conducteur. La conduction peut être améliorée grâce à un dopage convenable. Quelques propriétés des polymères conducteurs sont citées dans le paragraphe relatif aux composés conjugués. Bibliographie [1] Manuel d'expériences de chimie - UNESCO Société chimique de France - Université de Montpellier.
[19] Diphénylacétylène by I. D. Campbell and G. Eglinton.
[40] P. Cadiot; W. Chodkiewicz, Chemistry of Acetylenes; Viehe, H. G., Ed.; Marcel Dekker: New York, 1969; pp 597-647. A. Streitwieser Jr, C. H. Heathcock - Introduction to Organic Chemistry, Macmillan Publishing Co.
Vous pouvez, si vous le souhaitez, utiliser le contenu de cette page dans un but pédagogique et non commercial. Texte, dessins, photographies : Gérard Dupuis - Lycée Faidherbe de LILLE
Les principales caractéristiques de la molécule d'ozone ont été vues dans le chapitre consacré aux composés éthyléniques.
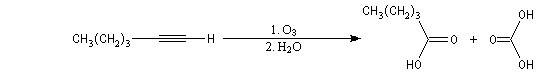
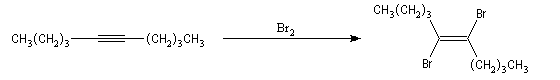
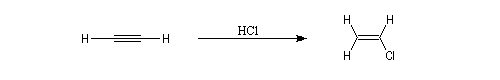
La réaction ressemble à celle qu'on observe avec les alcènes mais les alcynes sont un peu moins réactifs. La régiosélectivité suit la règle de Markovnikov.
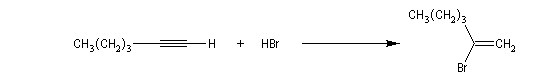
Une deuxième molécule de HBr s'additionne sur le composé précédent.
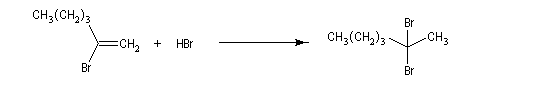
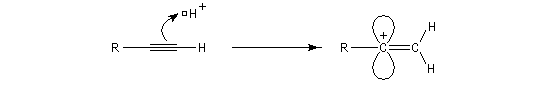
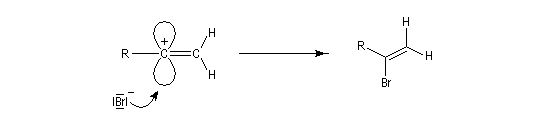
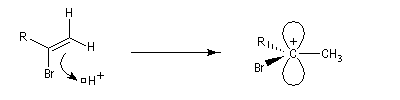
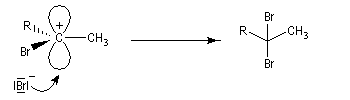
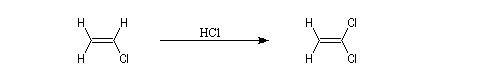
L'addition d'eau sur les alcynes est catalysée par les ions Hg2+. A partir d'un alcyne terminal on obtient un aldéhyde. En particulier l'éthyne (acétylène) , conduit à l'éthanal.
Cette réaction qui fut longtemps à la base du procédé industrielle de fabrication de l'éthanal n'est plus utilisée à cause du coût élevé de la production de l'acétylène. Actuellement, l'éthanal est synthétisé
à partir de l'éthène grâce au procédé Wacker beaucoup plus économique.
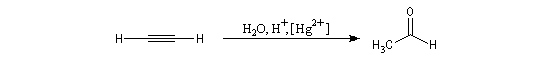
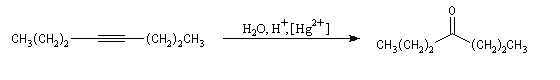
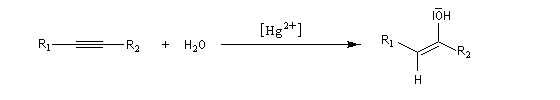
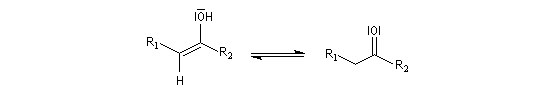
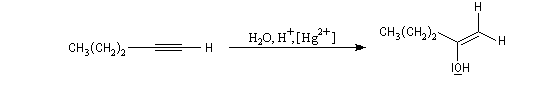
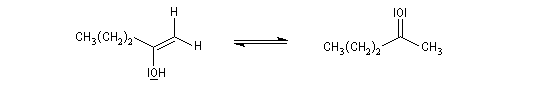
Quelques propriétés des hydrures de bore ont été vues dans le chapitre consacré aux composés éthyléniques. L'addition sur la triple liaison s'effectue avec une régiosélectivité inverse de celle obtenue quand la règle de Markovnikov s'applique.
L'addition de BH3 sur un alcyne conduit à l'addition sur les deux liaisons p.
En présence de boranes assez encombrés comme le dicyclohexylborane, ou le 9-BBN, l'hydroboration des alcynes terminaux conduit aux 1-alcénylboranes avec une excellente régiosélectivité.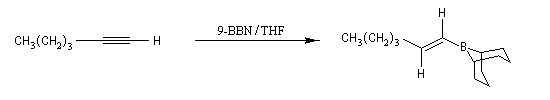
L'oxydation des 1-alcénylboranes permet la préparation d'aldéhydes car le produit d'oxydation est un énol. 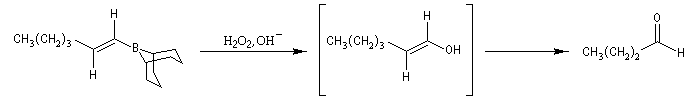

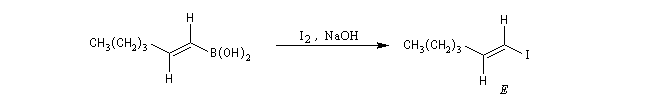
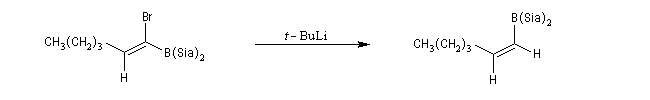
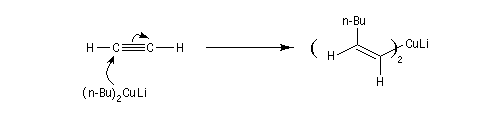
L'addition d'un cuprate lithien sur un alcyne permet de passer à un organocuprate vinylique (réactif de Normant) [41].
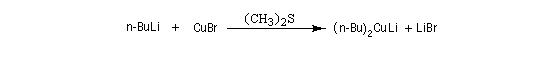
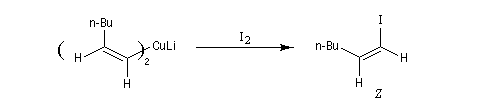
Réactif de Schwartz (à compléter)
En présence d'initiateurs de radicaux la régiosélectivité est l'inverse de celle prévue par la règle de règle de Markovnikov. La réaction suit une régiosélectivité comparable à l'effet l'effet Karrasch observé chez les alcènes.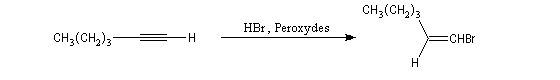
Les allènes sont des diènes possédant deux doubles liaisons consécutives.
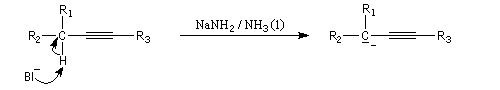
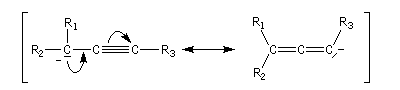
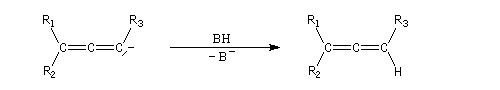
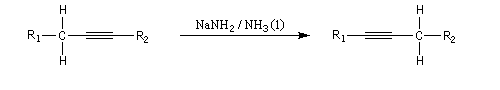
La trimérisation de l'éthyne conduit au benzène.
La tétramérisation de l'éthyne conduit au cyclooctatétraène.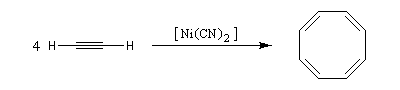
La première synthèse du polyacétylène (PA) a été réalisée par le chimiste japonais H. Shirakawa en 1971 en utilisant un catalyseur de Ziegler et Natta. A basse température, on obtient le
polymère entièrement cis. H. Shirakawa a obtenu le prix Nobel de chimie en 2000 [29].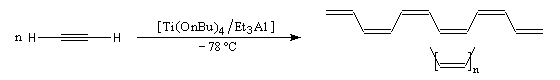
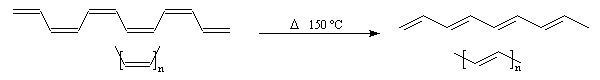
[2] P. Rendle, M. V. Vokins, P. Davis, Experimental Chemistry (Edward Arnold).
[3] L. F. Fieser, K. L. Williamson - Organic Experiments (D. C. Heath and Company).
[4] R. Adams, J. R. Johnson, C. F. Wilcox - Laboratory Experiments in Organic Chemistry (The Macmillan Company, Collier-Macmillan Limited).
[5] G. K. Helmkamp, H. W. Johnson Jr, Selected Experiments in Organic Chemistry (W. H. Freeman and Co).
[6] Vogel's Textbook of Practical Organic Chemistry (Longman).
[7] J. R. Mohrig, D. C Neckers, Laboratory Experiments in Organic Chemistry (D. Van Nostrand Company).
[8] R. Q. Brewster, C. A. Van der Werf, W. E. Mc Ewen, Unitized Experiments in Organic Chemistry (D. Van Nostrand Company).
[9] F. G. Mann, B. C. Saunders, Practical Organic Chemistry (Longman).
[10] M. T. Yip, D. R. Dalton, Organic Chemistry in the Laboratory (D. Van Nostrand Company).
[11] Miller, Neuzil, Modern Experimental Organic Chemistry (DC Heath, 1982).
[20] [18]-annulene by K. Stöckel and F. Sondheimer
[21] Trimethylsilylacetylene by Andrew B. Holmes and Chris N. Sporikou
[22] 1,2,3-triphenylcyclopropenium bromide by Ruo Xu and Ronald Breslow
[23] 1,4-bis (trimethylsilyl)Buta-1,3-diyne by Graham E. Jones, David A. Kendrick, and Andrew B. Holmes
[24] Ethyl-(Z)-crotonate by Michael J. Taschner, Terry Rosen, and Clayton H. Heathcock.
[25] 1-acetylcyclohexanol by Gardner W. Stacy and Richard A. Mikulec.
[26] Histrionicotoxin by Neil Edwards and Mark Reed University of Sussex at Brighton
[27] Les alcynes Ronald W. Rinehart
[28] 1,4-bis(trimethylsilyl)buta-1,3-diyne by Graham E. Jones, David A. Kendrick, and Andrew B. Holmes
[29] The Nobel Prize in Chemistry 2000
[30] (Z)-1-iodohexene by A. Alexakis, G. Cahiez, and J. F. Normant.
[31] SYNTHESIS OF TERMINAL 1,3-DIYNES VIA SONOGASHIRA COUPLING by Amanda L. Kohnen and Rick L. Danheiser.
[40] Y. Rubin, M. Kahr, C. B. Knobler, F. Diederich, C. L. Wilkins, J. Am. Chem. Soc. 1991, 113, 495-500.
[41] Normant, J (1971). "Synthese stereospecifique and reactivite d' organocuivreux vinyliques". Tetrahedron Letters 12 : 2583
[42]Sonogashira, K.; Tohda, Y.; Hagihara, N. “A Convenient Synthesis of Acetylenes: Catalytic Substitutions of Acetylenic Hydrogen with Bromoalkenes, Iodoarenes, and Bromopyridines.” Tetrahedron Lett., 1975, 50, 4467-4470.
J. March - Advanced organic chemistry, Wiley Interscience.
F. A. Carey, R. S. Sunberg - Advanced Organic Chemistry, Plenum Press 1990.
V. Minkine - Théorie de la structure moléculaire, Editions Mir 1979.
N. T. Ahn - Introduction à la chimie moléculaire, Ellipses 1994.
R. Brückner - Mécanismes réactionnels en chimie organique, De Boeck Université 1999.
J. C. Chottard, J. C. Depezay, J. P Leroux - Chimie fondamentale, Hermann 1982.
P. Laszlo - Logique de la synthèse organique. Cours de l'Ecole Polytechnique, Ellipses, 1993.
P. Atkins - Molecular Quantum Mechanics, Oxford University Press 1983.
J. -L. Rivail - Eléments de chimie quantique à l'usage des chimistes, InterEditions du CNRS, 1989.
décembre 2011