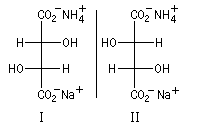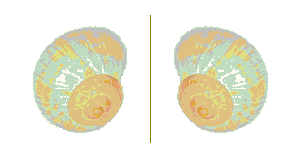
Cours de chimie Organique. G. Dupuis - Lycée Faidherbe de Lille
Chiralité et énantiomérie
Définitions
La constitution d'une entité moléculaire précise la nature et les modes d'union des atomes formant cette entité, en incluant la multiplicité des liaisons sans tenir compte de leur disposition dans l'espace.
La configuration d'une entité moléculaire est la disposition spatiale des atomes ou des groupes d'atomes de cette entité. Ce terme est propre aux stéréo-isomères dont l'isomérie n'est pas due à des différences de conformations.
L'étude structurale des stéréoisomères est l'objet de la stéréochimie, qu'on appelait autrefois la chimie dans l'espace et dont l'origine remonte aux travaux du chimiste hollandais J. H. van't Hoff et du chimiste français J. A. Le Bel qui émirent indépendamment en 1874 l'hypothèse du carbone tétraédrique. Van't Hoff fut le premier lauréat du prix Nobel de chimie en 1901 pour ses travaux concernant les équilibres chimiques et la pression osmotique [4].
Chiralité
Un objet chiral, en particulier une entité moléculaire chirale, n'est pas superposable à son image dans un miroir plan. C'est le cas de chacune des coquilles d'escargots représentées ci-dessous.
La chimie de coordination fournit de nombreux exemples de complexes chiraux impliquant des atomes variés qui ne sont pas nécessairement tétracoordinés :
Une approche historique du concept de chiralité est développée dans la conférence prononcée en 1975 par Vladimir Prelog lors de la remise de son prix Nobel. Elle est intitulée "La Chiralité en Chimie" [1].
Enantiomérie
L'objet chiral et son image, elle-même chirale, sont appelés énantiomères. On nomme énantiomérie la relation entre ces deux structures. Notons que les énantiomères étaient souvent appelés naguère des inverses optiques en raison de leurs pouvoirs rotatoires spécifiques opposés.
Deux stéréoisomères non énantiomères sont appelés diastéréo-isomères.
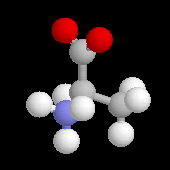
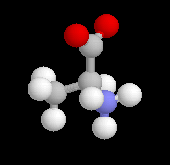
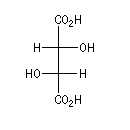
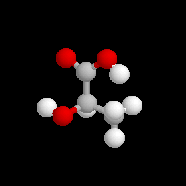
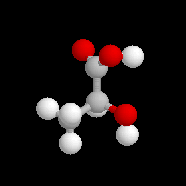
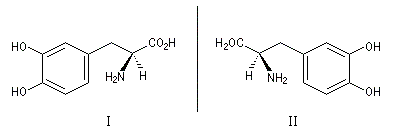
L'alanine naturelle est la molécule représentée sur l'image de droite (II). On sait synthétiser une autre molécule d'alanine représentée à gauche (I). Ces deux molécules sont images l'une de l'autre dans un miroir plan et non superposables. Ce sont donc des énantiomères.
|
|
|
L'alanine naturelle est la molécule représentée sur l'image de droite. On l'appelle L-alanine dans la nomenclature de Fischer. L'alanine naturelle est un composé dextrogyre caractérisé par un pouvoir rotatoire spécifique : |
Ces énantiomères sont représentés ci-dessous en utilisant le mode de représentation de Cram.
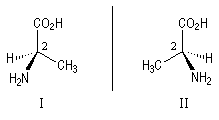
La configuration absolue d'une entité moléculaire chirale ou d'un groupe chiral est la disposition spatiale des atomes ou des groupes d'atomes qui distingue cette entité ou ce groupe de son image dans un miroir. Les configurations absolues des énantiomères de l'alanine permettent de les distinguer au moyen des stéréodescripteurs R et S, obtenus par application des règles de Cahn, Ingold, Prelog.
N > C1(CO2H) > C3 (CH3) > H
|
Molécule |
I |
II |
|
Configuration absolue |
R |
S |
L'alanine naturelle est donc l'acide (+)-(2S)-2-aminopropanoïque.
Un autre exemple intéressant d'un point de vue historique est celui des sels de sodium et d'ammonium de l'acide tartrique séparés par Pasteur en 1848.
Opérations de symétrie|
Elément de symétrie |
S1 |
S2 |
Sn |
|
Equivalence |
plan (s) |
centre (i) |
axe inverse (S) |
|
|
|
Les molécules (I) et (II) sont chirales et énantiomères l'une de l'autre. Elles possèdent un axe de symétrie C2. 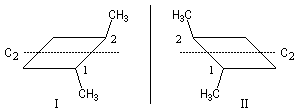 |
Ces dernières années, de nombreuses molécules de symétrie C2 ont été utilisées comme ligands dans la préparation de catalyseurs chiraux permettant des synthèses énantiosélectives. Le premier exemple de ce type, dû au chimiste français H. Kagan en 1971 est le ligand DIOP obtenu à partir d'acide tartrique chiral. Un autre exemple est le ligand BINAP de Noyori.
Exemples
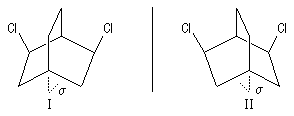
|
|
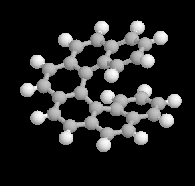
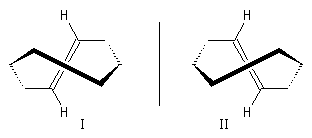
Les molécules I et II sont images l'une de l'autre dans un miroir et superposables. Elles sont achirales car elles possèdent un plan de symétrie passant par le pont. |
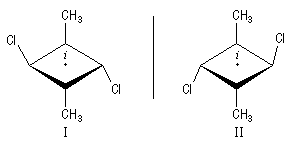
|
|
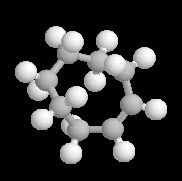
Les molécules I et II sont images l'une de l'autre dans un miroir et superposables. Elles sont achirales car elles possèdent un centre de symétrie au milieu du cycle. On passe de I à II en tournant la structure autour d'un axe passant par le centre du cycle. |
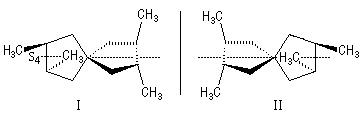
|
|
Les molécules I et II sont images l'une de l'autre dans un miroir et superposables. Elles sont achirales car elles possèdent un axe inverse de symétrie S4 passant par l'atome de carbone central et le milieu de chaque cycle. On passe de I à II en tournant de 90° autour de cet axe. |
Certaines conformations d'une molécule de configuration donnée peuvent être chirales. La conformation synclinale du butane, considérée comme une structure figée, est chirale. Les structures I et II sont donc énantiomères.
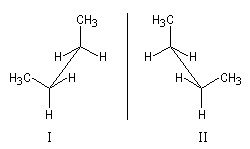
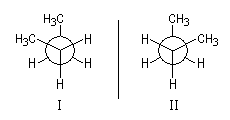
Ces structures ont la même énergie potentielle microscopique. La barrière d'énergie qui les sépare est relativement faible et, à la température ordinaire, les populations moyennes des molécules I et II sont égales. Dans ces conditions, les propriétés du butane à l'échelle macroscopique sont celles d'une molécule achirale.
Pour qu'une molécule soit configurationnellement chirale, il faut que toutes ses conformations le soient.
D'autres exemples de cette situation sont fournis par l'acide mésotartrique ainsi que le (cis)-1,2-diméthylcyclohexane et la cis-décaline.
Molécules possédant un centre chiral
Définition
On appelle centre chiral un atome maintenant un ensemble d'atomes ou de groupes d'atomes dans une disposition non superposable à son image dans un miroir. Cette notion de centre chiral est très générale et ne se limite pas aux composés du carbone.
Par exemple, dans le complexe Tris(éthylènediamine)cobalt (III) entre le Co (III) et l'éthylènediamine : [Co(en)3]2+, l'atome de cobalt est un centre chiral.
Atome de carbone asymétrique
Un exemple classique de centre chiral est celui d'un atome de carbone relié à quatre groupes différents. Il est appelé, traditionnellement, atome de carbone asymétrique. L'appellation carbone asymétrique, introduite par van't Hoff fait référence aux structures comportant un atome de carbone de ce type, qui ne possèdent aucun élément de symétrie et qui sont donc asymétriques. Un atome asymétrique, centre chiral, avec ses substituants, constitue un exemple de groupe stéréogène car il peut être considéré comme à l'origine d'une stéréo-isomérie.
|
|
|
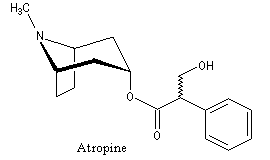
Les images ci-contre représentent les deux énantiomères de l'acide tropique. L'atropine , un alcaloïde extrait de la belladone, est un mélange racémique de dérivés des acides tropiques. Ce composé possède la remarquable propriété de dilater la pupille de l'œil. Il était utilisé au XVII ème siècle par les femmes italiennes dans le but d'augmenter la profondeur de leur regard. De là vient le nom de belladonne : bella dona qui signifie jolie femme en italien . |
Les formules développées de ces énantiomères sont dessinées ci-dessous en utilisant le mode de représentation de Cram.
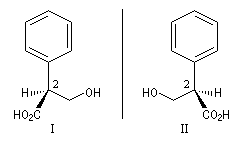
Les configurations absolues des énantiomères possèdant un atome de carbone asymétrique sont nommées de façon systématique en utilisant les règles de Cahn, Ingold, Prelog.
C1 (CO2H) > C3 (CH2OH) > C(Ph) > H
|
Molécule |
I |
II |
|
Configuration absolue |
S |
R |
Le composé I sera nommé : acide (2S)-2-phényl-3-hydroxypropanoïque.
Retenons que l'existence d'un atome de carbone asymétrique, et un seul, dans une molécule est une condition suffisante de chiralité. Ce n'est pas une condition nécessaire. Les allènes, les spirannes, constituent des exemples de molécules chirales sans atome de carbone asymétrique.
Atome d'azote
Les amines tertiaires substituées par des groupes différents, sont chirales. A la température ordinaire, les deux énantiomères ne peuvent généralement pas être séparés en raison de l'inversion de configuration rapide de l'atome d'azote.
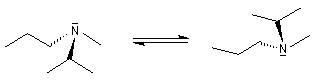
La réaction entre une amine tertiaire et un dérivé halogéné est appelée alkylation d'Hofmann. Elle fournit un ion ammonium quaternaire qui est chiral si les quatre groupes sont différents. On peut alors, théoriquement au moins, séparer les deux énantiomères.
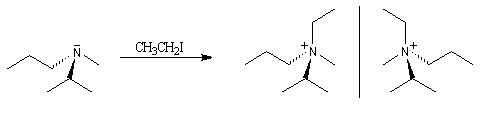
Dans certains cas, l'inversion de l'atome d'azote peut être ralentie ou même bloquée par suite d'une configuration particulière de la molécule. C'est le cas quand l'atome d'azote est engagé dans un système ponté ou à l'intérieur d'un petit cycle comme dans les aziridines substituées à l'azote
|
|
La base de Tröger est un composé polycyclique dans lequel deux atomes d'azote forment un pont. La rigidité de ce système polycyclique empêche l'inversion de l'atome d'azote. Le mélange racémique des deux énantiomères de ce composé a été dédoublé par le chimiste suisse d'origine croate V. Prelog en 1944 par chromatographie sur phase stationnaire chirale [26] . |
Atome de phosphore
Les phosphanes substitués par des groupements différents sont chiraux. La hauteur de la barrière d'interconversion est généralement comprise
entre 125 et 145 kJ.mol-1. Elle est suffisante pour qu'on puisse séparer les énantiomères à la température ordinaire. L'inversion
pyramidale n'a lieu qu'à une température plus élevée.
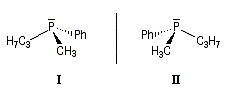
C'est en utilisant une phosphine chirale de ce type que le chimiste américain W. S. Knowles a préparé en 1968 un catalyseur chiral permettant la première hydrogénation énantiosélective d'un dérivé du styrène.
Atome de soufre
Les sulfoxydes présentent des barrières d'interconversion encore plus élevée, comprise entre 145 et 185 kJ.mol-1. Le mélange racémique peut être dédoublé en énantiomères à la température ordinaire.
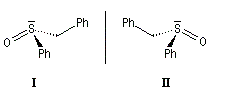
Molécules contenant plusieurs centres chiraux
Lorsqu'il y a plusieurs atomes de carbone asymétriques dans la molécule, celle-ci peut être chirale ou bien achirale car la chiralité est une propriété globale de la structure. Le cas des acides tartriques (2R, 3R) et (2S, 3S) d'une part et (2R, 3S) d'autre part, constituent des exemples de cette situation.
Composés méso
On appelle composé méso (du grec mesos : milieu), un composé comportant deux centres chiraux de configurations absolues opposées. Il s'agit d'un composé achiral. Son pouvoir rotatoire spécifique est donc nul.
Certains époxydes qui possèdent un plan de symétrie appartiennent aussi à cette catégorie. C'est le cas pour le composé ci-dessous.
|
|
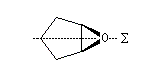 |
L'ouverture des époxydes méso peut être rendue énantiosélective de différentes manières. En voici deux :
|
|
|
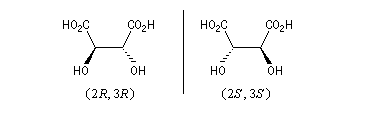
Acides (2R, 3R)-tartrique et (2S, 3S)-tartrique
|
|
|
Il existe deux acides tartriques chiraux :
|
Ces composés sont dessinés ci-dessous en utilisant la représentation de Cram.
Une représentation utile est la projection de Fischer. Il faut faire attention que contrairement à ce que pourrait laisser croire un examen rapide de ces projections, il n'existe pas de centre de symétrie dans ces molécules. En effet la chaîne carbonée n'est pas plane mais cambrée vers l'arrière.
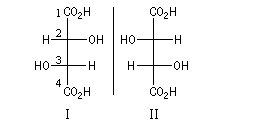
Les configurations absolues sont données par les règles de Cahn, Ingold, Prelog. Ces molécules constituent le couple de configuration relative like (R*R*).
|
Molécule |
I |
II |
|
Configuration absolue |
(2R, 3R) |
(2S, 3S) |
Les acides (2R, 3R) et (2S, 3S) tartriques sont largement utilisés dans la préparation de nombreux réactifs chiraux. Ils constituent une source de chiralité à la fois pratique et de coût modique. Un exemple est la préparation du ligand DIOP d'un complexe au ruthénium utilisé comme catalyseur d'hydrogénation énantiosélective.
Acide (2R, 3S)-tartrique
Bien qu'il y ait deux atomes de carbone asymétriques dans leur molécule, il existe trois et non quatre acides tartriques car les configurations (2R, 3S) et (2S, 3R) sont les mêmes :
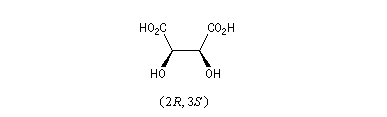
|
|
L'acide (2R, 3S)-2, 3-dihydroxybutane-1,4-dioïque encore appelé acide mésotartrique est un diastéréo-isomère des acides (2R, 3R) et (2S, 3S) tartriques. On peut obtenir ses sels par traitement des acides (2R, 3R) et (2S, 3S) tartriques par une base forte à chaud. |
La molécule d'acide (2R, 3S)-tartrique, possède une conformation dans laquelle on trouve un plan de symétrie S situé entre les atomes de carbone 2 et 3. Ce plan apparaît nettement sur la représentation de Cram de la conformation éclipsée (B) ou sur la projection de Fischer (C) de la molécule.
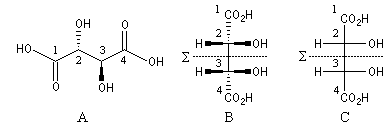
Notons qu'il existe des conformations chirales de l'acide (2R, 3S)-tartrique. Ces conformations possèdent la même énergie potentielle microscopique et elles sont donc en quantités égales. Les propriétés macroscopiques de l'acide (2R, 3S)-tartrique sont celles d'une molécule achirale.
Molécules possédant un axe de chiralité
Allènes
Les allènes sont des diènes cumulés. Ils possèdent un atome de carbone lié par deux liaisons doubles à deux autres atomes de carbone contigus. Les substituants des atomes extrêmes sont situés dans des plans P et P' perpendiculaires. Les molécules I et II sont énantiomères.
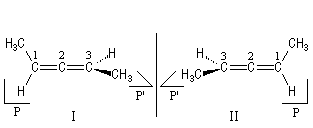
La nomenclature de la chiralité axiale utilise les stéréodescripteurs a-R et a-S.
|
|
|
La chiralité des allènes dissymétriques avait été prévue par van't Hoff bien avant que la première séparation effective de deux énantiomères soit réalisée.
|
La première séparation effective de deux énantiomères de ce type a été réalisée par les chimistes anglais P. Maitland et W. H. Mills dans le cas décrit ci-dessous. Les molécules sont représentées en projection le long de l'axe des liaisons doubles [25].
Notons que l'existence de la chiralité axiale n'implique pas nécessairement que la molécule soit asymétrique puisqu'il peut exister un axe de symétrie C2 suivant la bissectrice de l'angle entre les plans des laisons. Lorsque les atomes portés par les atomes de carbone terminaux sont différents, les molécules sont asymétriques (appartenance au groupe C1).
Lorsque deux groupes portés par les atomes de carbone extrèmes sont identiques, la molécule est achirale car elle possède alors un plan de symétrie.
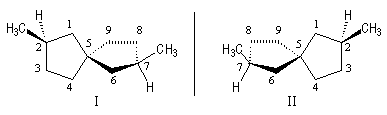
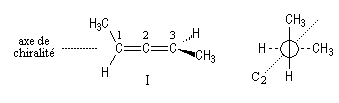
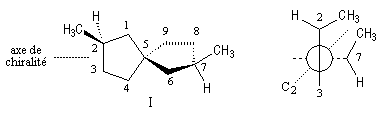
On a représenté ci-dessous la molécule en utilisant une projection de Newman suivant l'axe de chiralité.
Un spiranne achiral possédant un axe de symétrie inverse S4 est donné plus haut.
Atropisomérie
L'atropisomérie constitue un exemple d'énantiomérie lié à l'empêchement de la rotation autour d'une liaison simple. Le mot atropisomérie est formé à partir des mots grecs tropein, tourner et meros, partie. Les molécules I et II sont énantiomères.
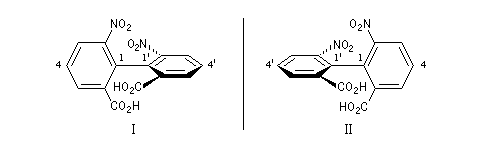
La molécule I est représentée à droite en utilisant une projection de Newman suivant l'axe de chiralité. Notons que, bien qu'étant chirale, la structure conserve un axe de symétrie C2 (à ne pas confondre avec l'axe de chiralité.)
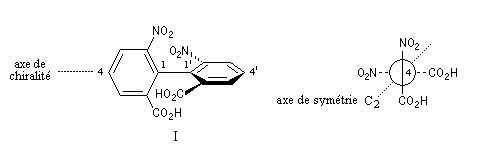
L'encombrement stérique des groupes nitro et acide est tel qu'à la température ordinaire la vitesse d'interconversion est suffisamment faible pour qu'on puisse séparer les deux énantiomères.
|
|
|
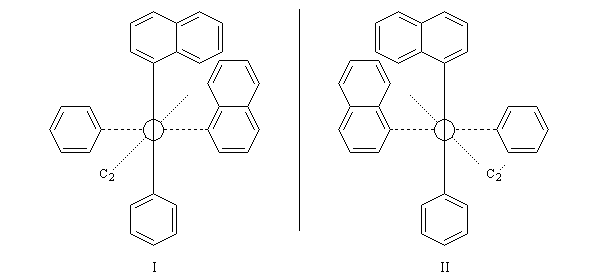
Dans le cas de biphényles, on passe d'un énantiomère à l'autre par rotation autour de la liaison entre les deux groupes phényles. Selon l'encombrement stérique des groupes fixés sur les cycles, la vitesse d'interconversion est plus ou moins élevée à une température donnée. Le chimiste japonais Okï a défini un critère de séparabilité des espèces énantiopures en partant de l'idée qu'elles doivent avoir des temps de demi-vie d’au moins 103 secondes pour pouvoir être séparées.
Du point de vue synthétique, ces composés peuvent être obtenus par la réaction de couplage d'Ullman. Plus récemment, les réactions de couplage croisé au palladium telles que la réaction de Negishi ou celle de Suzuki ont été mises à profit.
Certains atropisomères comme les binaphtyles atropiques sont utilisés comme ligands chiraux du ruthénium dans la synthèse de catalyseurs d'hydrogénation asymétriques en phase homogène. Ces catalyseurs permettent des synthèses asymétriques énantiosélectives avec des excès énantiomériques dépassant 95 %.
De nombreuses molécules naturelles contiennent dans leur structure des stéréoisomères de ce type. A titre d'exemple, les problèmes soulevés par la synthèse totale de la vancomycine, un glycopeptide tricyclique (surnommé l'antibiotique de la dernière chance) ont récemment montré l'intérêt des méthodes de couplage telles que la réaction de Suzuki dans les synthèses stéréosélectives des isomères atropiques. Les solutions apportées par les groupes de D. A. Evans (Université de Harvard) et K. C. Nicolaou (Institut de recherche Scripps) figurent parmi les résultats les plus remarquables obtenus en synthèse organique ces dernières années.
Molécules possédant un plan de chiralité
Un plan de chiralité peut être considéré comme résultant de la désymétrisation d'un plan de symétrie par suite d'une ou plusieurs modifications sur la molécule comme par exemple une substitution.
Cyclènes
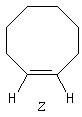
Les cyclènes sont des composés éthyléniques de formule brute CnH2n-2. Le (E)-cyclooctène est le plus petit des cyclènes trans. C'est un composé dans lequel la liaison double impose une contrainte importante et une certaine rigidité. L'interconversion entre les énantiomères I et II est rendue difficile car la molécule doit passer par une conformation particulièrement contrainte. Il a été préparé en 1953 par A. C. Cope en mettant à profit la réaction d'élimination d'Hofmann [6].
L'interconversion est suffisamment lente à la température ordinaire pour qu'on puisse séparer les énantiomères. Elle ne devient notable qu'à partir de 120 °C.
|
|
|
|
|
|
|
Le (E)-cyclodécène possède un cycle plus grand que le (E)-cyclooctène et il est par conséquent moins rigide. L'interconversion entre les deux énantiomères est rapide dès la température ordinaire puisque la demi-vie de racémisation est de l'ordre de 5 min à 0 °C.
Stéréochimie des métallocènes
Les métallocènes constituent un groupe important dont le représentant le plus connu est le ferrocène.
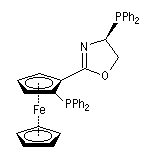
Hélicité
Les hélicènes sont des molécules hélicoïdales constituées de noyaux benzéniques accolés par un côté. Des hélices de différentes longueurs ont été synthétisées.
|
|
L'hexahélicène est un enchaînement ouvert de six cycles benzéniques accolés par un côté. La molécule adopte une conformation non plane à cause de la répulsion entre les cycles situés aux deux extrémités de la molécule. |
Les énantiomères correspondent aux deux sens d'enroulement de l'hélice :
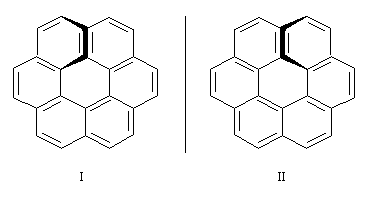
Ces énantiomères ont été effectivement séparés par les chimistes américains M. S. Newman et D. Lednicer en 1956. Ils se caractérisent par un pouvoir rotatoire spécifique exceptionnel [a] = 3700 °.g-1.dm.cm3.
Cette propriété remarquable a permis à H. Kagan de l'Université d'Orsay, de réaliser la première synthèse asymétrique d'hélicènes chiraux, induite par la lumière polarisée, en 1971. Le pouvoir rotatoire est très élevé bien que le rendement de la réaction soit très faible [22].
La chiralité liée à l'hélicité est un phénomène très général qu'on retrouve dans de nombreux polymères d'origine biologique comme les protéines et les acides nucléiques comme l'ADN.
Distinction entre conformations et configurations
Propriétés physiques comparées des énantiomères
En revanche des énantiomères ont des propriétés différentes vis à vis d'un phénomène physique dissymétrique. En particulier les pouvoirs rotatoires spécifiques des énantiomères sont opposés.
Le mélange équimolaire de deux énantiomères s'appelle mélange racémique.
On donne ci-dessous quelques propriétés des acides (+)-(2R, 3R)-2,3-dihydroxybutanedioïque et (-)-(2S, 3S)-2,3-dihydroxybutanedioïque énantiomères ou acides tartriques et de leur mélange racémique.
|
Composé |
TF (°C) |
Densité d |
Solubilité (g/100 g) |
[a] (°.dm-1.g-1.cm3) |
|
(2R, 3R)-tartrique |
170 |
1,76 |
147 |
+12 |
|
(2S, 3S)-tartrique |
170 |
1,76 |
147 |
-12 |
|
mélange racémique |
205 |
1,68 |
25 |
0 |
Mélanges racémiques
Généralités
Le mélange équimolaire de deux énantiomères s'appelle mélange racémique. Les propriétés physiques d'un mélange racémique sont souvent complètement différentes de celles des énantiomères purs. Les pouvoirs rotatoires spécifiques de deux énantiomères sont égaux en valeur absolue mais de signes opposés. De ce fait, le pouvoir rotatoire spécifique d'un mélange racémique est nul par compensation. Le passage d'un composé énantiopur au mélange des deux énantiomères en quantités égales s'appelle racémisation.
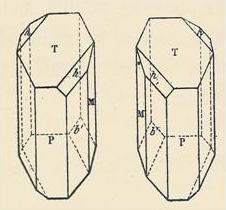 |
Figure de gauche : cristaux de tartrate de sodium et d'ammonium énantiomères séparés par Pasteur. Les molécules correspondantes sont représentées ci-dessous en utiisant la projection de Fischer. La mesure du pouvoir rotatoire spécifique de chaque composé pur, montre que le premier est dextrogyre et le second est lévogyre. |
 |
Le mot racémique vient du latin racemus qui signifie grappe de raisin. L'acide (+)-(2R,3R)-tartrique se trouve à l'état naturel dans le jus de raisin et c'est l'observation de la cristallisation de sels de cet acide (tartrates) dans des barriques de vin d'Alsace qui est à l'origine du travail de Pasteur. |
|
|
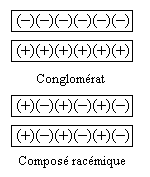
On distingue deux types de racémiques :
|
Le passage d'un mélange à l'autre fait intervenir la température. Dans l'exemple cité plus haut, le mélange racémique des sels d'ammonium et de sodium des acides tartriques (+) et (-) est un composé racémique au dessus de 28 °C. Alors, les cristaux ne sont plus constitués de molécules énantiopures mais comme on l'a dit plus haut de cristaux constitués de quantités égales des deux énantiomères. Dans un tel cas, la méthode de Pasteur n'est plus utilisable.
La distinction entre composé racémique et conglomérat racémique est particulièrement nette lorsqu'on observe les diagrammes binaires pour chacun d'eux.
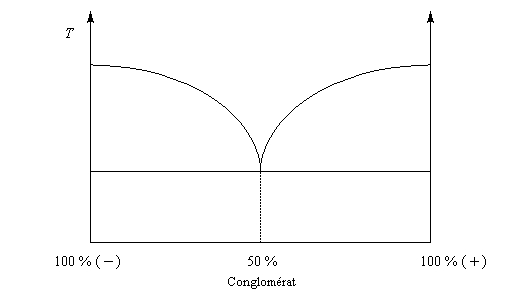
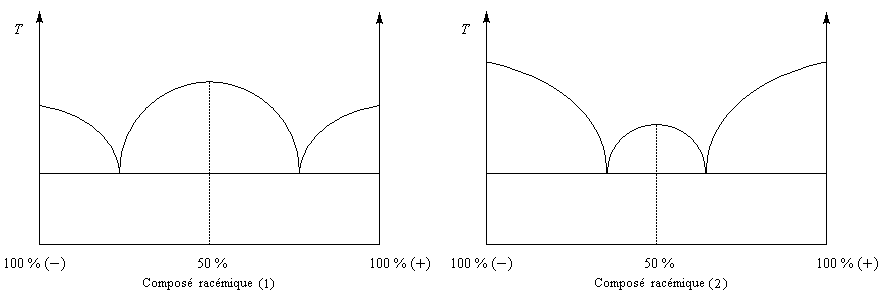
|
|
Les fruits de la plante Datura représentée sur la photo ci-contre, contiennent de très nombreux alcaloïdes. Parmi ceux-ci, l'atropine est le nom donné au mélange racémique de deux énantiomères : la (+)-hyoscyamine et la (-)-hyoscyamine.
|
Dédoublement par formation de diastéréo-isomères
Une méthode générale de dédoublement d'un mélange racémique consiste à effectuer une réaction entre les deux énantiomères du mélange et un agent résolvant chiral. Il s'agit souvent de molécules d'origine naturelle appartenant à ce qu'il est convenu d'appeler le fond chiral (en anglais : chiral pool.) On obtient ainsi deux composés diastéréo-isomères dont les propriétés physico-chimiques sont différentes. La réaction ne doit évidemment pas modifier la stéréochimie des centres, des axes ou des plans de chiralité.
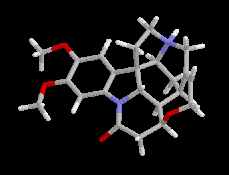
Lorsque les énantiomères possèdent une fonction basique on utilise des agents résolvants acides comme les acides (+)-tartrique et (-)-tartrique. Lorsque les énantiomères possèdent une fonction acide, on utilise des amines chirales comme la brucine ou la strychnine qui existent à l'état naturel. L'inversion de l'atome d'azote de ces composés est rendue impossible du fait de l'existence d'un système ponté rigide à l'exemple de la base de Tröger. Les structures de la brucine et de la strychnine sont données ci-dessous.
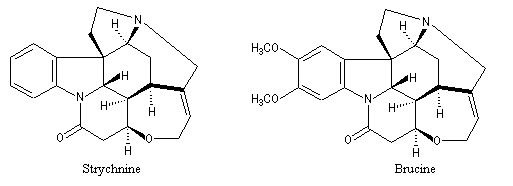
|
|
|
 |
|
C'est en faisant réagir la cinchonine naturelle (un seul énantiomère), et les acides (R, R) et (S, S) tartrique, que Pasteur parvint en 1853 a séparer les sels diastéréo-isomères par cristallisation fractionnée. Les acides tartriques énantiomères sont ensuite obtenus facilement par acidification du mélange. Le principe d'une telle séparation est donné ci-dessous. Dans le cas d'un mélange racémique d'acides AH opposés à une base B.
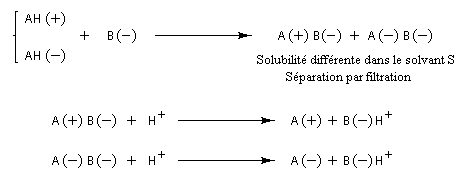
L'exemple ci-dessous concerne le dédoublement de l'acide lactique racémique par la (S)-brucine symbolisée par (S)-B.
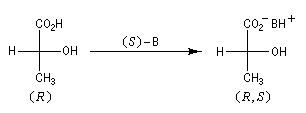
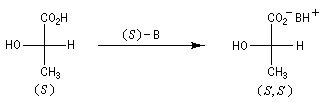
Là encore, un traitement acide de chacun des sels diastéréoisomères permet de récupérer les acides lactiques.
Lorsque les énantiomères à séparer possèdent des propriétés basiques, on utilise un acide chiral facile a obtenir énantiopur tel que l'acide (+)-tartrique [5].
Dédoublement par chromatographie sur phase stationnaire chirale
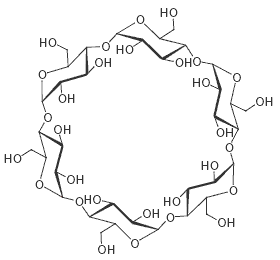
Cette technique, qui peut être utilisée à l'échelle industrielle, consiste à utiliser la chromatographie liquide haute performance (HPLC) avec une phase stationnaire chirale. Les cyclodextrines sont fréquemment utilisées. Ces composés sont des molécules cycliques qu'on peut considérer comme des oligomères du glucose. L'a-cyclodextrine, représentée ci-dessous, comporte 6 unités de D-glucose, la b-cyclodextrine en comporte 7 et la g-cyclodextrine 8.
Il s'agit d'une structure chirale. La molécule se présente sous la forme d'une cavité dont la géométrie est en tronc de cône ce qui lui permet de jouer le rôle de molécule-hôte. On notera que groupes hydroxyles polaires sont plutôt situés à l'extérieur de cette cavité tandis que l'extérieur de celle-ci comporte des groupes hydrophobes. Deux molécules chirales énantiomères forment avec leur hôte des complexes diastéréo-isomères. Les constantes d'équilibre entre la molécule hôte et la molécule invitée sont donc différentes, ce qui permet de les séparer.
|
|
|
Dédoublement cinétique
Le dédoublement cinétique (en anglais : kinetic resolution KR) est basé sur la différence de vitesse de réaction de chaque énantiomère vis à vis d'un même agent (réactif ou catalyseur) chiral. Les états de transition correspondants
sont des diastéréo-isomères qui possèdent des énergies différentes. Cette question est étudiée plus en détail dans le chapitre consacré à la stéréochimie dynamique.
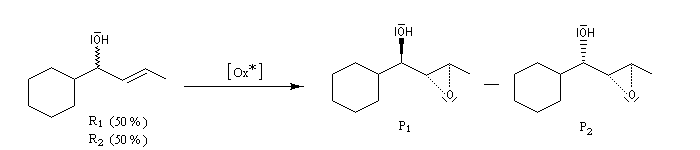
Dans l'exemple précédent, basé sur une époxydation de Sharpless, les composés R
Lorsqu'un équilibre de racémisation suffisamment rapide peut s'établir entre les deux énantiomères, il est possible de convertir théoriquement tout le mélange racémique en un seul énantiomère. Il s'agit dans ce cas de dédoublement cinétique dynamique (en anglais : dynamic kinetic resolution DKR).
Dédoublement enzymatique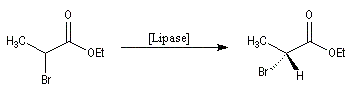
Cette méthode a été utilisée avec succès par Pasteur dès 1848 pour séparer les énantiomères de l'acide tartrique au moyen de levure de bière.
Ces sujet sont aussi abordés dans le chapitre consacré à la stéréochimie dynamique.
Propriétés chimiques comparées des énantiomères
|
|
|
L'acide (2R)-lactique est l'acide (2R)-2-hydroxypropanoïque, représenté sur l'image de gauche. Il est lévogyre (-). Ce composé a été découvert par Scheele dans le lait laissé longtemps à l'air (on dit que "le lait est tourné"). Son énantiomère, l'acide (2S)-lactique (image de droite) est dextrogyre (+). On le trouve dans le sang. Son accumulation dans les muscles, chez les sportifs soumis à un long effort est à l'origine des crampes. La polymérisation de l'acide lactique fournit un polymère biodégradable, le PLA. |
En milieu biologique, l'acide (2S)-2-hydroxypropanoïque ou (2S)-lactique peut être oxydé en acide 2-oxopropanoïque ou acide pyruvique par la forme oxydée du nicotinamide adénine-dinucléotide NAD+ grâce à la présence d'une molécule chirale : l'enzyme lactate déshydrogénase (LDH).
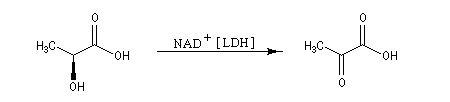
Son énantiomère, l'acide (2R)-2-hydroxypropanoïque ou (2R)-lactique A', dans les mêmes conditions, n'est pas oxydé. Il s'agit d'un exemple de réaction énantiosélective.
Les mécanismes de la perception des odeurs ne sont pas parfaitement élucidés. On constate expérimentalement que des énantiomères ont des odeurs différentes.
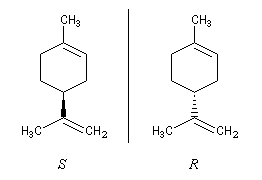
Il en est de même des récepteurs du goût.
|
|
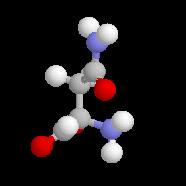
La (S)-asparagine (I) représentée ci-contre, se trouve à l'état naturel dans les jeunes asperges auxquelles elle confère une saveur amère caractéristique. A. Piutti découvrit en 1886 que son énantiomère d'origine synthétique, la (R)-asparagine (II), possède un goût sucré. |
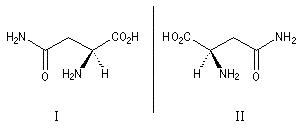
Propriétés pharmacologiques des énantiomères
Les propriétés pharmacologiques de deux énantiomères peuvent être très différentes. Lorsqu'on est certain de l'innocuité de l'énantiomère d'une molécule active, le médicament est prescrit sous forme racémique. L'énantiomère actif est appelé eutomère tandis que celui qui n'a pas les propriétés recherchées est qualifié de distomère. Un distomère peut être une molécule inactive donc inutile. Mais il peut aussi arriver qu'il s'agisse d'un composé toxique. L'exemple de la thalidomide prescrite sous forme racémique dans les années 60 est resté tristement célèbre. De nos jours, chaque année, environ 70 % des nouvelles autorisations de mise sur le marché portent sur des composés énantiopurs.
Notons qu'il existe des cas où bien que le principe actif soit constitué de l'un des énantiomères pur, une racémisation intervient in-vivo.
|
|
Il existe deux molécules de propanolol énantiomères. Leurs effets physiologiques sont différents :
|
Les médicaments sont de plus en plus souvent commercialisés sous forme énantiopure. En 2002, le marché total des molécules énantiopures était estimé à environ 150 milliards de dollars. Près de 80 % de ce marché concerne des molécules à but pharmaceutique.
Le Naproxène ou acide 6-méthoxy-a-méthyl-2-naphthalèneacétique est obtenu dans l'industrie pharmaceutique sous forme racémique par synthèse non stéréosélective. Seul l'énantiomère (S)-(+) possède des propriétés anti-inflammatoires. Il est donc nécessaire d'isoler cet énantiomère du mélange racémique. Le naproxène étant un acide, on utilise un agent de dédoublement avec lequel les deux énantiomères forment des sels diastéréo-isomères. En l'occurence, on utilise la N-propylglucosamine, une amine chirale qui peut être obtenue sous forme énantiopure.
De plus en plus de médicaments chiraux sont obtenus par synthèse stéréosélective.
Un précurseur de la dopamine, utilisé comme traitement dans la maladie de Parkinson, existe sous deux formes énantiomères. Seul l'énantiomère (I), appelé L-DOPA a une activité thérapeutique, l'autre est toxique. Il est obtenu par hydrogénation énantiosélective à partir de la vaniline selon un procédé mis au point par W. S. Knowles (prix Nobel 2001) pour Mosanto.
.
Phéromones
Les phéromones (du grec phrein : transférer et horman : exciter) sont secrétées par les insectes. Leur activité biologique est généralement reliée à une stéréochimie bien définie.
La disparlure est une phéromone d'attraction sexuelle de la mite gitane (gypsy moth) représentée ci-dessous :
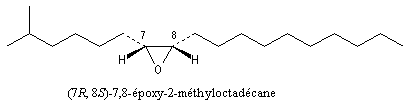
|
|
Le (+)-(7R, 8S)-7,8-époxy-2-méthyloctadécane ou disparlure est une phéromone sexuelle du Bombyx disparate. Sa structure a été établie par Beroza et coll. en 1970, en concentrant le produit issu de plusieurs dizaines de milliers d'insectes. Seul l'énantiomère (+) est actif. L'énantiomère (-) n'a aucune action biochimique même si sa concentration est 106 fois plus grande que celle de l'énantiomère (+). La synthèse de cette molécule a constitué l'une des premières grandes applications de l'époxydation de Sharpless. |
Le premier exemple d'extraction puis d'identification d'une phéromone d'insecte est celle du bombykol, une phéromone sexuelle du Bombyx du mûrier par A. F. J. Butenandt en 1959 [3].
Depuis cette date, de nombreuses phéromones ont été étudiées et synthétisées. La synthèse asymétrique énantiosélective de la frontaline a été réalisée de plusieurs manières. L'une d'entre-elles met à profit la dihydroxylation de Sharpless.
La frontaline est une phéromone d'agrégation de certains coléoptères appartenant à la famille
des scolitidae. Parmi ces insectes, le scarabée Dendroctonus frontalis Zimmermann (Southern Pine Beetle) est l'insecte le plus destructeur de forêts de pins dans le sud des USA.
L'élucidation de la structure des phéromones et leur synthèse sont à l'origine d'une méthode élégante de contrôle des populations d'insectes. Ce contrôle est très important car certains ravagent des cultures, d'autres transmettent des maladies. La technique consiste à attirer les insectes mâles avec une phéromone synthétique et les détourner ainsi des femelles. Cette méthode entraîne beaucoup moins d'effets secondaires que l'utilisation des insecticides classiques.
Bibliographie
Ouvrages théoriques
Baltimore Lectures on Molecular Dynamics and the Wave Theory of Light, C. J. Clay and Sons, Cambridge University Press Warehouse, London 1904
Stereochemistry of Organic Compounds, Ernest L. Eliel and Samuel H. Wilen, Wiley, New York, 1994
J. March, Advanced organic chemistry, Wiley Interscience.
F. A. Carey, R.J. Sundberg, Advanced organic chemistry, 3d edition, Plenum Press, 1990.
H. Kagan, La stéréochimie organique, PUF, 1975.
J. L Pierre, Principes de stéréochimie organique statique, A. Colin, 1971.
A. Kirmann, J. Cantacuzène, P. Duhamel - Chimie organique T 1, A. Colin, 1971.
K. Mislow, Introduction to stereochemistry, W. A Benjamin, New York, 1966.
J. Jacques, La molécule et son double, Hachette, 1992.
J. Jacques, A. Collet, S. H. Wilen, Enantiomers Racemates and Resolutions, Wiley-Intersciences, New York, 1981.
Alexander von Zelewsky, Stereochemistry of Coordination Compounds, Wiley, 1996, New York.
Articles sur la toile
[1] Chirality in Chemistry by V. Prelog
[2] E. Fischer, The Nobel Prize in Chemistry 1902
[3] A. Butenandt, The Nobel Prize in Chemistry 1939
[4] J. H. van't Hoff The Nobel Prize in Chemistry 1901
[5] R(+)- and S(-)-phenylethylamine by Addison Ault.
[6] trans-Cyclooctene by Arthur C. Cope and Robert D. Bach
[7] Lipase Katalysed kinetic resolution of alcohols via chloroacetate esters by A. Schwartz, P. Madan, J. K. Whitesell, and R. M. Lawrence.
[8] Chirality and odour perception
[9] Termes utilisés en stéréochimie Commission générale de terminologie et de néologie (CNRS)
[10] Cours de stéréochimie de J. C. Gressier
[11] Basic Terminology of Stereochemistry IUPAC Recommendations 1996
[12] The Importance of Chirality Gareth J. Rowlands
[13] Synthèse totale de la strychnine
[14] La Chimie Organique dans la Conception des Médicaments et dans leurs Modes d’Action par Stéphane Quideau, Université de Bordeaux.
[15] Diastereoisomerism Application à la séparation d'énantiomères by Gareth J. Rowlands
[16] The Nobel Prize in Chemistry 1913 Alfred Werner
Articles
[20] Van't Hoff, Le Bel Centenial, O. B. Ramsay, J. American Chemical Society, Washington D. C., 1975.
[21] A. Collet - Chiralité et médicaments, Revue du Palais de la Découverte 237 p 23 (1996).
[22] A. Moradpour, J.-F. Nicoud, G. Balavoine, H. B. Kagan, Asymetric Synthesis of hexahelicen. J. Am. Soc.,1971, 93, 2353.
[23] G. H. Christie, J. Kenner, J. Chem. Soc. 1922, 121, 614-620.
[24] Dang, T. P. & Kagan, H. B. (1971) J. Chem. Soc. Chem. Commun., 48.
[25] P. Maitland, W. H. Mills, Nature, 1935, 135, 994.
[26] V. Prelog, P. Wieland (1944). "Über die Spaltung der Tröger'schen Base in optische Antipoden, ein Beitrag zur Stereochemie des dreiwertigen Stickstoffs". Helvetica Chimica Acta 27 (1): 1127–1134
[27] M.S. Newman, D. Lednicer, The synthesis and resolution of Hexahelicen, 1956, 78, 4765-4770.
[28] R. B. Woodward, Michael P. Cava, W. D. Ollis, A. Hunger, H. U. Daeniker, K. Schenker (1954). "The total synthesis of strychnine". J. Am. Soc.,76: 4749.
Gérard Dupuis - Lycée Faidherbe - LILLE
juillet 2018