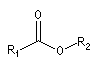
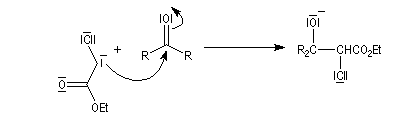
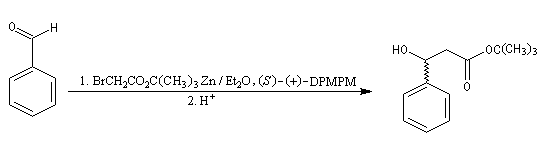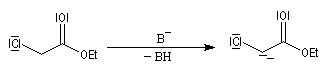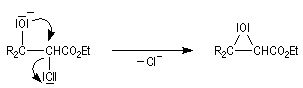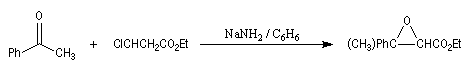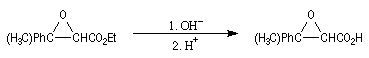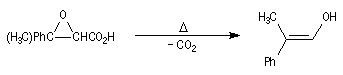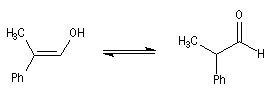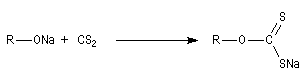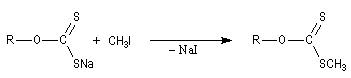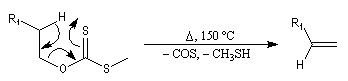|
|
L'acétate de vinyle est préparé par oxyacétylation de l'éthène en utilisant l'acide d'acide éthanoïque et l'oxygène de l'air. La réaction est effectuée
entre 150 °C et 200 °C en utilisant comme catalyseur un alliage de palladium et d'or en présence d'acétates alcalins.
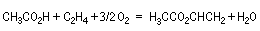 Il sert à préparer le polyacétate de vinyle qui intervient :
Il sert à préparer le polyacétate de vinyle qui intervient :
- dans les peintures latex [31] ;
- qui permet de synthétiser les alcools polyvinyliques.
|
Propriétés physiques
Constantes physiques
Les esters de faible masse molaire sont des composés volatils. Leurs températures d'ébullition sont légèrement plus élevées que celles des hydrocarbures de masse
masse molaire comparable. Cela s'explique par l'existence d'interactions dipôle-dipôle chez les esters qui n'existent pas
chez les hydrocarbures. En revanche les températures d'ébullition sont beaucoup plus faibles que chez les acides carboxyliques.
|
Formule |
Nom |
TE (°C) |
|
HCO2CH3 |
méthanoate de méthyle |
32 |
|
CH3CO2CH3 |
éthanoate de méthyle |
57 |
|
CH3CO2CH2CH3 |
éthanoate d'éthyle |
77 |
|
CH3CO2(CH2)2CH3 |
éthanoate de n-propyle |
102 |
Comme beaucoup d'esters ont une odeur fruitée agréable ils interviennent souvent comme composants volatils de parfums.
Structure des esters
Il est intéressant de comparer la longueur de la liaison C-Z dans
un éther et dans un ester.
|
Groupe Z |
d (C-Z) (nm) éther |
d (C-Z) (nm) ester |
|
-OCH3 |
0,143 |
0,136 |
Le raccourcissement observé quand on passe d'un éther à un ester s'interprète par la méthode de la mésomérie. Dans l'une des formes mésomères, la liaison C-Z est double.
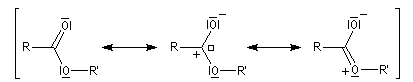
Les formes mésomères précédentes montrent que l'atome de carbone du groupe fonctionnel des esters est moins électrophile que celui des composés carbonylés. On interprète ainsi le fait que
les esters sont moins réactifs dans les additions nucléophiles que les composés carbonylés.
Spectroscopie
Spectroscopie infrarouge
Il est intéressant de comparer les absorptions du groupement carbonyle des cétones, des esters et des anhydrides :
|
Groupe fonctionnel |
cétones |
esters |
|
s (cm-1) |
1705 - 1725 |
1735 - 1750 |
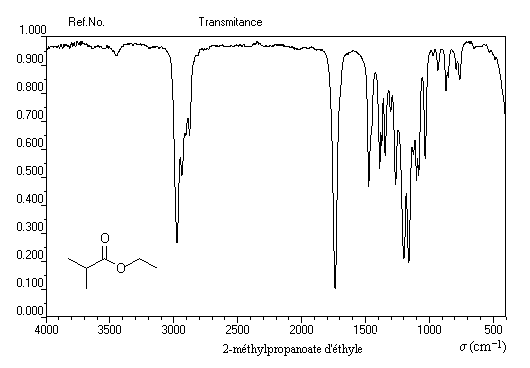
Les esters présentent deux absorptions caractéristiques :
- le groupe carbonyle vers 1740 cm-1 ;
- le vibrateur -C-O-C- vers 1235 cm-1.
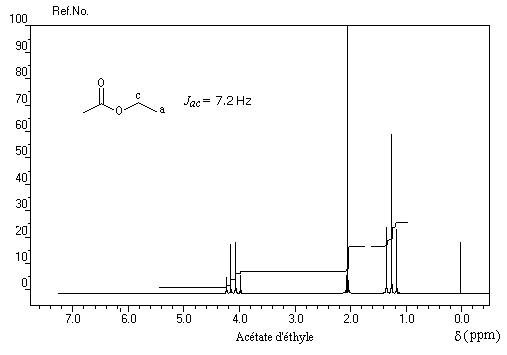
Spectroscopie de RMN
Les protons portés par des atomes de carbone liés à une fonction ester résonnent pour des champs plus faibles que chez les cétones. Le tableau suivant permet de comparer le déplacement chimique des protons cas de dérivés méthylés.
|
Groupe |
CH3-CO- |
CH3-O-CO- |
|
d (ppm) |
2 |
4 |
Réduction des esters
Réduction en alcools par LiAlH4
LiAlH4 réduit les esters en alcools. Pour l'exemple suivant, voir [6].
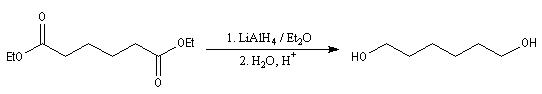
Réduction par le sodium
La réaction de Bouveault et Blanc, est connue depuis longtemps (1903). Elle consiste à réduire les esters en alcools par le sodium dans un solvant protogène comme l'éthanol.
L'exemple suivant concerne la préparation de l'alcool laurylique [38] :
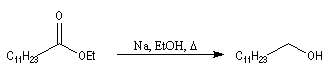
Le mécanisme de cette réaction est le suivant :
- transfert d'un électron du sodium vers le groupe carbonyle ;
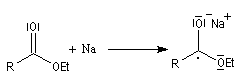
- protonation de l'intermédiaire ;
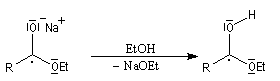
- transfert d'un deuxième électron ;
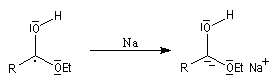
- protonation par le solvant qui conduit à l'alcool.
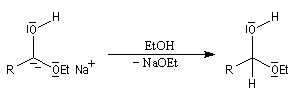
Depuis l'avènement des méthodes mettant en jeu les hydrures complexes, comme le DIBAL-H, la réaction de Bouveault et Blanc n'est employée que dans certains cas très particuliers de réductions chimiosélectives d'hydroxy-esters.
Réaction d'Adkins
La réaction d'Adkins est l'hydrogénolyse des esters par le dihydrogène en présence de Cu(Cr2O3) comme catalyseur. Cette réaction qui exige une pression et une température élevées, est surtout utilisée à l'échelle industrielle.
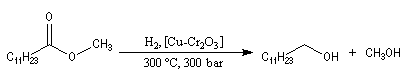
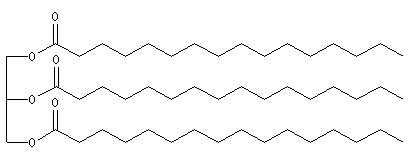
L'hydrogénolyse des triglycérides constitue une méthode de préparation des alcools à longue chaîne carbonée. L'exemple suivant concerne la tripalmitine.
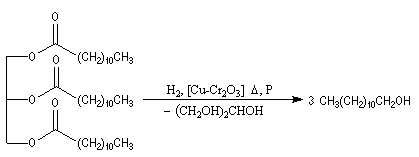
Hydrogénation catalytique
L'hydrogénation des liaisons éthyléniques des esters d'acides gras insaturés est effectuée en présence de dihydrogène, à chaud, le nickel servant de catalyseur. La trioléine peut ainsi être réduite en tristéarine. L'hydrogénation des huiles
végétales contenant des triglycérydes d'acide gras insaturés, est à la base de la préparation industrielle de la margarine.
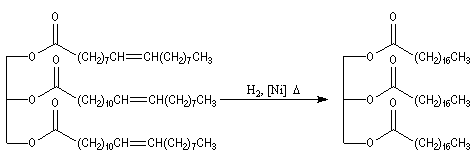
Réduction par le DIBAL-H
Les esters sont réduits en aldéhydes par un hydrure neutre d'aluminium, le bis (2-méthylpropyl)-aluminium Al(i-Bu)2H (diisobutylaluminium : DIBAL-H) dans un solvant apolaire comme le toluène à basse température.
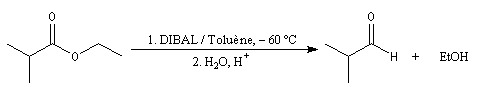
La réaction suivante permet la synthèse de l'aldéhyde de Garner, substrat très utile dans la synthèse stéréosélective des sucres [46].
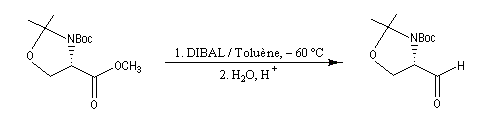
En utilisant 2 moles de réactif par mole d'ester, la réduction va jusqu'au stade de l'alcool.
Les lactones sont réduites en lactols.
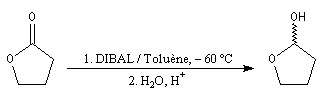
Dans une telle molécule, on reconnaît une fonction aldéhyde masquée sous forme d'hémiacétal. Sous cette forme la réduction de la fonction aldéhyde ne peut intervenir. Elle est ainsi protégée.
En présence de catalyseurs comme les ions H+, il est possible de déplacer l'équilibre vers la forme ouverte.
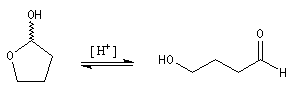
|
|
Le bis (2-méthylpropyl)-aluminium Al(i-Bu)2H (diisobutylaluminium ou DIBAL-H) est un hydrure neutre encombré. On le prépare par la réaction suivante :
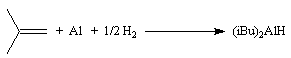
Il est beaucoup plus facile à manipuler que LiAlH4. Sa chimiosélectivité est différente de celle des hydrures chargés comme NaBH4. Il faut noter la présence d'une orbitale vide sur l'aluminium ce qui confère au réactif un caractère d'acide de Lewis. Le mécanisme de réduction est d'ailleurs différent de celui des hydrures précédents [49].
|
Le produit de réduction des esters par le DIBAL-H dépend du solvant :
- à - 60 °C, dans un solvant apolaire, l'intermédiaire organoaluminique formé est stable jusqu'à l'hydrolyse. On obtient l'aldéhyde ;
- à température ambiante, dans un solvant polaire comme le THF, l'intermédiaire subit une élimination qui conduit à l'aldéhyde. Celui-ci réagit avec le DIBAL-H plus rapidement que l'ester et la réaction conduit finalement à l'alcool.
Le mécanisme implique la formation d'un complexe acide-base de Lewis entre l'hydrure métallique agissant comme un accepteur et le composé carbonylé donneur.
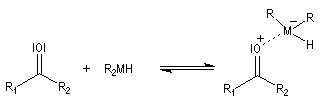
Le transfert de l'ion hydrure s'effectue de façon intramoléculaire au sein du complexe.
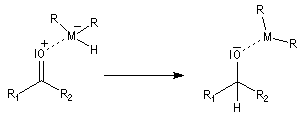
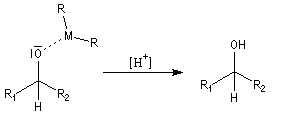
Une hydrolyse permet d'obtenir le produit final.
Condensation acyloïne des esters
Lorsqu'on traite un ester en présence de sodium dans un solvant aprotique comme le toluène, on observe le couplage des radicaux intermédiaires. L'absence de solvant protique ne permet pas la réaction de réduction de Bouveault et Blanc.
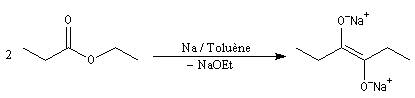
Le traitement du composé obtenu par l'acide acétique conduit à un énol.
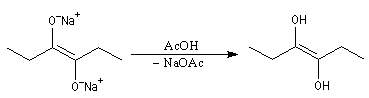
L'énol se tautomérise en a-hydroxycétone ou acyloïne.
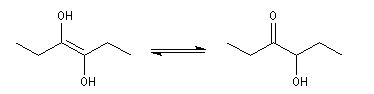
Le mécanisme admis pour cette réaction est le suivant :
- l'ester accepte un électron du sodium ce qui conduit à la formation d'un radical anion ;

- le couplage de deux radicaux fournit un dianion ;
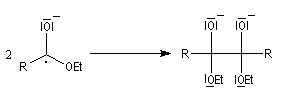
- le dianion subit une élimination pour conduire à un composé dicarbonylé 1,2 ; la force motrice de cette réaction est la grande énergie de liaison du groupe carbonyle ;
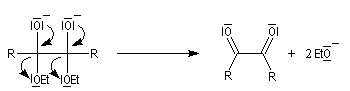
- le composé dicarbonylé accepte à son tour deux électrons du métal ;
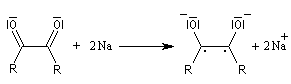
- par couplage on obtient cette fois la base conjuguée d'un énol ;
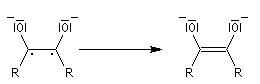
- le traitement par un acide comme l'acide acétique fournit l'énol qui, par tautomérie, conduit au produit final.
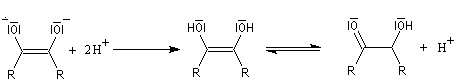
Cette réaction est à rapprocher de la réduction duplicative des composés carbonylés. Elle permet
la synthèse de composés à grands cycles. Cela est illustré par la synthèse de la 2-hydroxycyclodécanone [26].
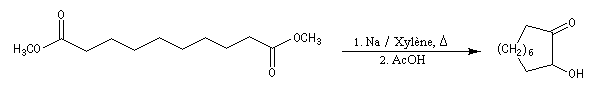
La réduction par le zinc de cette acyloïne fournit la cyclodécanone [27].
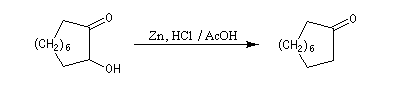
La condensation acyloïne des esters a été appliquée à la synthèse des cétones à grand cycle comme la civétone ou la muscone (réaction ci-dessous.)
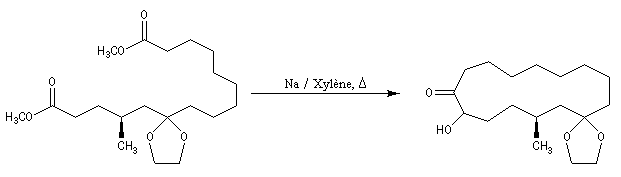
Le succès de cette réaction est attribué au fait que le couplage s'effectue à la surface du métal. On notera qu'il est nécessaire d'utiliser de grandes dilutions dans ce type de synthèse afin de limiter les réactions intermoléculaires.
Réactions avec les organomagnésiens
Esters
La préparation des organomagnésiens a été vue dans le chapitre consacré aux composés organométalliques. La réaction entre un ester et un organomagnésien, conduit à un intermédiaire tétraédrique instable car l'ion éthanolate est un nucléofuge moyen.
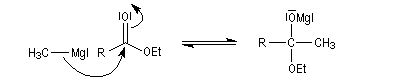
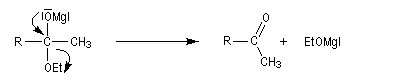
La cétone qui provient de la fragmentation de cet intermédiaire réagit plus vite avec l'organométallique que l'ester de départ. On obtient donc un alcool tertiaire dans lequel on retrouve deux fois la chaîne carbonée du magnésien.
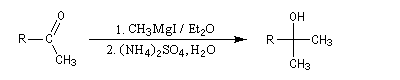
Un exemple d'application est la synthèse du triphénylméthanol. Notons que cet alcool tertiaire est facilement identifiable grâce au carbocation triphénylméthyle auquel il conduit en milieu acide.
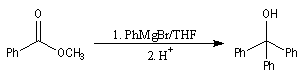
W. H. Perkin utilise cette méthode dans sa synthèse historique du terpinéol (1904).
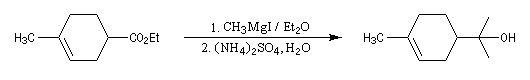
On peut cependant préparer des cétones à partir des esters en opérant dans un solvant de forte constante diélectrique, très polarisant et très solvatant comme le HMPT. Dans ce solvant, la cétone est transformée en énol. La réaction subséquente avec l'organométallique n'a plus lieu.
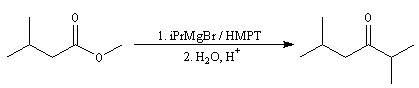
Orthoesters
On appelle orthoester un cétal d'ester. L'orthoformiate d'éthyle dérive de l'acide méthanoïque (acide formique). La réaction débute par l'élimination d'un ion éthanolate à partir de l'orthoester. Cette élimination est facilitée par la présence de l'organomagnésien qui joue le rôle d'acide de Lewis. On obtient un acétal.
Le traitement de l'acétal en milieu acide permet d'obtenir l'aldéhyde. L'exemple suivant concerne la synthèse de l'heptanal [44].
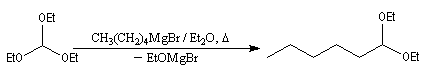
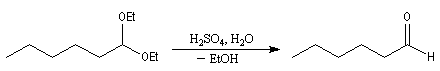
Hydrolyse des esters
Propriétés générales
L'hydrolyse d'un ester est la réaction inverse de la estérification de Fischer. Elle conduit à un acide carboxylique ou un ion carboxylate et à un alcool. L'état d'équilibre est atteint au bout d'un temps très long en l'absence de catalyse. Comme un catalyseur catalyse aussi bien la réaction directe
que la réaction inverse, on peut utiliser les mêmes catalyseurs que pour l'estérification. Le plus simple est l'ion H+.
- en milieu acide, on obtient l'acide carboxylique. La transformation conduit à un équilibre :
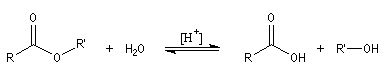
- en milieu basique, le produit final est l'ion carboxylate. Les ions OH- ne sont pas des catalyseurs car ils sont consommés dans la réaction. La transformation est totale :
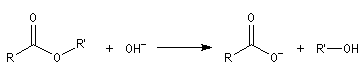
Milieu acide
L'hydrolyse des esters en milieu acide suit habituellement un mécanisme AAC2. Il s'agit de la réaction inverse de l'estérification de Fischer. Ce mécanisme a été vu dans le chapitre consacré aux alcools.
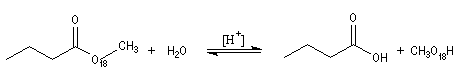
Certains esters comme le 2,4,6-triméthylbenzoate de méthyle ou mésitoate de méthyle sont hydrolysés en milieu acide selon un mécanisme AAC1.
Milieu basique
Le mécanisme habituel de l'hydrolyse basique des esters est de type BAC2. Les étapes sont les suivantes :
- addition de l'ion hydroxyde sur le groupe carboxyle de l'ester avec formation d'un intermédiaire tétraédrique. Cette étape bimoléculaire est cinétiquement déterminante. Comme les ions hydroxyde sont de bien meilleurs nucléophiles que l'eau, la saponification est plus rapide que l'hydrolyse ;
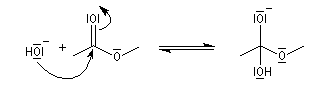
- fragmentation de l'intermédiaire tétraédrique avec élimination d'un ion éthanolate ;
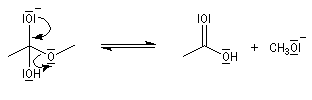
- déprotonation de l'acide carboxylique par l'ion éthanolate avec formation d'un ion carboxylate. Cette réaction quantitative rend la transformation globale totale.
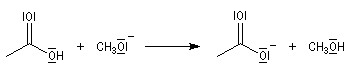
La saponification des triglycérides permet de préparer les sels alcalins des acides gras qui constituent les savons.
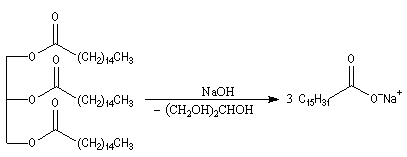
|
|
Le 2,4,6-triméthylbenzoate de méthyle ou mésitoate de méthyle est un dérivé de l'acide mésitoïque. Son hydrolyse ne peut s'effectuer selon
le mécanisme habituel d'hydrolyse des esters. La formation de l'intermédiaire tétraédrique est défavorisée pour des raisons stériques.
|
Certains esters comme le mésitoate de méthyle subissent une saponification en suivant un mécanisme inhabituel. En présence de soude concentrée, la réaction de substitution nucléophile de l'ion hydroxyde sur le méthyle de la fonction ester est beaucoup plus facile que l'addition sur le groupe carboxyle. La saponification suit un mécanisme BAL2.
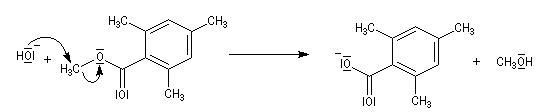
Transestérification
La réaction de transestérification constitue une méthode douce de préparation des esters. La transformation est équilibrée et l'équilibre peut être déplacé dans le sens
souhaité en utilisant un excès de réactif ou en retirant l'un des produits au fur et à mesure de sa formation.
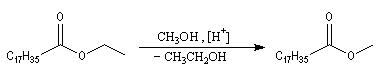
La réaction peut aussi avoir lieu de façon intramoléculaire [2].
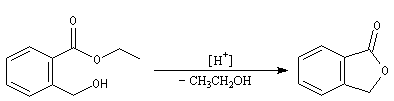
Par réaction de transestérification entre des triglycérides et un alcool comme le méthanol, on obtient des esters méthyliques d'acides gras et du glycérol. Ces esters sont utilisés comme carburants dans les moteurs Diesel. Ces carburants sont connus sous le nom générique de biodiesel [47].
Réactions avec l'ammoniac et les amines
Aminolyse des esters
La réaction entre une amine primaire ou secondaire et un ester constitue une méthode douce de préparation des amides.
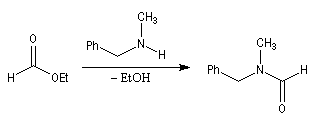
Ce type de réaction est utile pour préparer des amides cycliques à grands cycles ou macrolactames. On s'en sert également pour créer des liaisons amides à partir de composés difonctionnels. Un exemple de ce type est la synthèse des benzodiazépines [1,4].
Ammoniac
L'ammoniac réagit d'une façon comparable à celle des amines primaires. La réaction avec l'oxalate d'éthyle fournit l'oxamide [2].
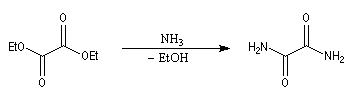
Enolates d'esters
Préparation
L'hydrogène en a de la fonction ester est acide avec un pK du couple (ester/énolate) de l'ordre de 25. L'acidité est donc plus faible que chez une cétone. Les ions énolate sont formés en traitant l'ester par une base très forte comme le LDA. Dans ce genre de transformation, on introduit l'ester dans la solution de LDA dans le THF de façon à éviter la réaction du carbanion formé sur
l'ester.
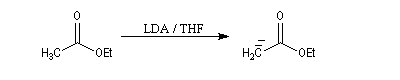
La stabilité de l'ion énolate obtenu peut être interprétée par la méthode de la résonance. Contrairement à ce qui se passe avec une cétone, il n'y a qu'un seul atome de carbone en a possible et le problème de la régiosélectivité ne se pose pas ici.
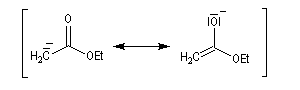
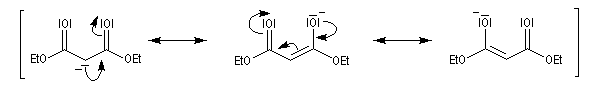
Chez un diester comme l'ester malonique l'acidité des atomes d'hydrogène du groupe méthylène est considérablement accrue et une base comme l'ion carbonate suffit à préparer l'énolate.
La stabilité de l'anion du malonate de diéthyle peut être interprétée par les formes mésomères suivantes.
Les ions éthanolate sont souvent utilisés bien que la réaction ne nécessite pas l'utilisation d'une base si forte car on ne risque pas de réaction concurrente de transestérification. Les ions hydroxyde ne sont pas utilisables car ils conduiraient
à la saponification de l'ester.
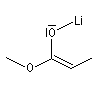
Formation d'énolates Z et E
L'obtention d'énolates Z et E à partir de composés carbonylés a été vue dans le chapitre correspondant. Compte-tenu de la méthode utilisée pour définir
les stéréodescripteurs Z et E, il faut faire attention qu'à une double liaison avec des substituants O-Li et alkyle en cis dans un énolate d'ester correspond à une configuration relative E alors que la même
relation cis autour de la double liaison d'un énolate de cétone correspond à une configuration Z.
|
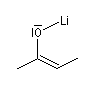
|
|
|
Enolate de cétone (Z) |
Enolate d'ester (E) |
Avec le LDA dans le THF, la formation de l'énolate E est favorisée.
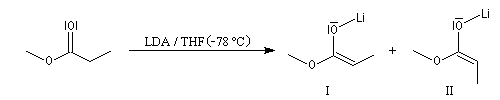
Alkylation des énolates
Les énolates d'esters peuvent être alkylés au moyen de dérivés halogénés. Dans la réaction suivante, le substrat est le bromure d'allyle très réactif.
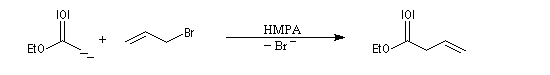
Dans ce type de réaction, la condensation de Claisen peut être une réaction secondaire génante. On s'en affranchit en ajoutant l'ester dans la solution de base dans le THF et non l'inverse.
L'alkylation d'énolates d'esters chiraux est à la base d'une méthode de synthèse diastéréosélective d'acides carboxyliques appelée synthèse de Helmchen.
Synthèse malonique
On cherche à passer d'un dérivé halogéné à un acide carboxylique possédant deux atomes de carbone de plus que le composé de départ.
Le réactif de départ est le propanedioate de diéthyle encore appelé malonate de diéthyle dont la préparation a été vue dans le chapitre sur les acides. La base la plus appropriée pour déprotoner le malonate de diéthyle est l'ion éthanolate. Bien
que cette réaction ne nécessite pas une base d'une telle force, l'avantage vient ici du fait que la
réaction concurrente de transestérification fournit le même produit que le réactif de départ.
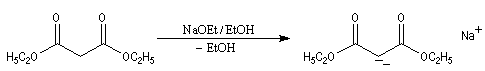
- L'alkylation de l'anion du diester est une substitution nucléophile sur le dérivé halogéné ;
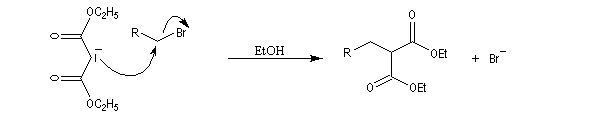
- le diester est ensuite hydrolysé, ce qui fournit le diacide ;
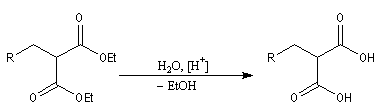
- la décarboxylation du diacide fournit le produit cherché.
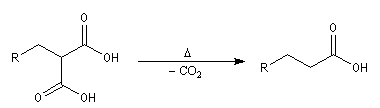
C'est par cette méthode que Perkin synthétisa le premier composé comportant un cycle cyclopropanique. L'exemple suivant est une variante de cette réaction qui met en jeu une catalyse par transfert de phase [12].
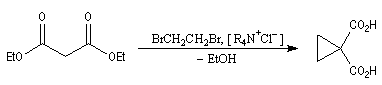
Un autre exemple est la synthèse de l'acide pélargonique, voir [34].
Variantes de la synthèse malonique
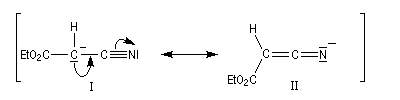
D'autres carbanions que ceux dérivant de diesters peuvent être utilisés. Dans l'exemple suivant, une fonction ester est remplacée par une fonction nitrile.
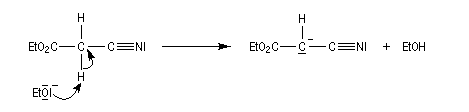
La méthode de la mésomérie permet de rendre compte de la stabilisation du carbanion.
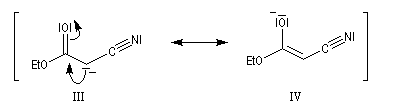
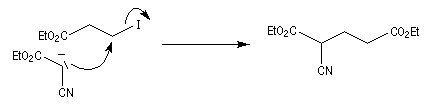
Une substitution nucléophile avec un dérivé halogéné comme substrat, permet de préparer le produit souhaité.
Synthèse acétylacétique
Le principe est le même que pour la réaction de synthèse malonique au lieu d'alkyler l'ester malonique il s'agit cette fois d'alkyler l'ester acétylacétique.
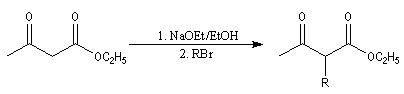
Le composé formé est un cétoester substitué. La synthèse peut alors être poursuivie selon deux voies :
- traité par une quantité catalytique d'éthanolate de sodium dans l'éthanol en excès, il subit une réaction de rétro-Claisen qui fournit deux esters ;
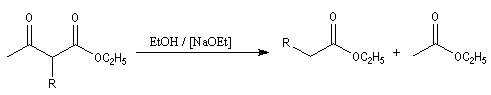
le cétoester substitué peut aussi être hydrolysé en b-cétoacide. La décarboxylation de ce dernier donne une cétone.
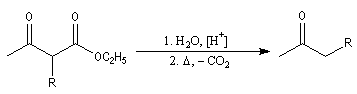
Réactions de condensation
Condensation de Claisen
La condensation de Claisen est une réaction de préparation des b-cétoesters. La transformation conduit à un état d'équilibre. La réaction inverse s'appelle rétro-Claisen.
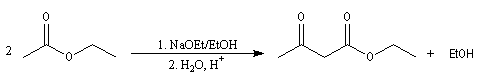
Pour cette réaction particulière, voir par exemple [11]. Pour des exemples récents d'application de cette réaction, voir [28].
Le mécanisme de cette réaction est le suivant :
- arrachage d'un atome d'hydrogène de l'ester par l'ion éthanolate jouant le rôle de base ;
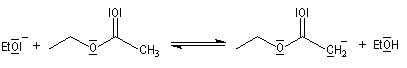
- addition nucléophile du carbanion de l'ester sur une deuxième molécule d'ester et formation d'un intermédiaire tétraédrique ;
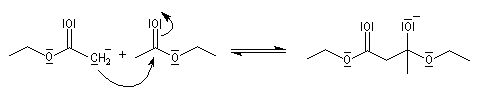
- élimination d'un ion éthanolate à partir de l'intermédiaire tétraédrique.
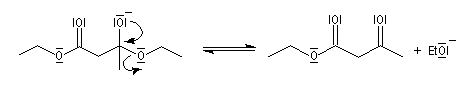
A priori une quantité catalytique d'éthanolate semble suffire. Cependant le b-cétoester obtenu est assez acide et le produit finalement obtenu est le sel formé entre ce composé et l'éthanolate. Il faut donc utiliser un équivalent
d'éthanolate de sodium par mole de produit formé.
Au lieu d'utiliser un énolate d'ester comme réactif nucléophile, on peut utiliser l'énolate d'un composé carbonylé. Les réactions suivantes peuvent être considérées comme des variantes de la réaction de Claisen
dans lesquelles l'ion énolate est formé à partir d'une cétone. Elles constituent des voies d'accès aux composés dicarbonylés 1, 3.
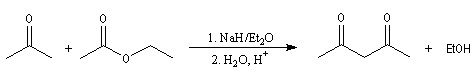
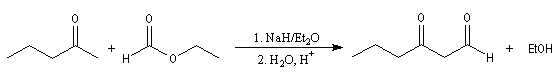
Un autre exemple est celui de la synthèse de l'acide pimélique ; voir [39].
Cyclisation de Dieckmann
Il s'agit d'un cas particulier de condensation de Claisen réalisée de façon intramoléculaire. Comme une condensation de Claisen ordinaire, elle conduit à un équilibre et peut être renversée. Elle est utilisable
pour synthétiser des cycles à 5, 6 ou 7 chaînons.
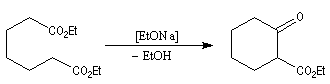
Voir par exemple [29].
La cyclisation de Dieckmann a été utilisée dans de nombreuses synthèses. La transformation ci-dessous est une étape dans la synthèse de l'équilénine par Bachmann en 1939 [52]. Cette molécule est la première hormone sexuelle à avoir été synthétisée.
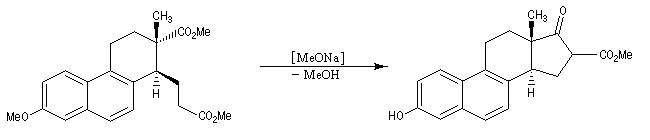
Le b-cétoacide est ensuite facilement décarboxylé pour conduire au produit.
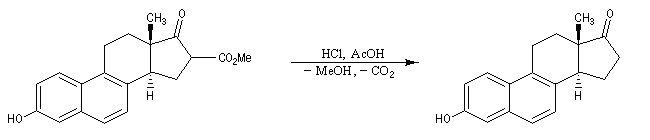
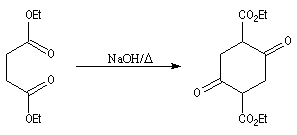
La réaction de Dieckmann n'est pas utilisable pour la synthèse de cycles contraints comme des cyclopropanes substitués. La transformation suivante,
fait intervenir une condensation de Claisen intermoléculaire préalable suivie d'une cyclisation de Dieckmann. Elle conduit à un cycle à six chaînons alors que la cyclisation
directe fournissant un squelette cyclopropanique n'est pas observée.
Condensation de Knoevenagel
La réaction de Knoevenagel est une réaction très générale entre un composé à méthylène actif et un aldéhyde ou une cétone. La plupart du temps, le composé carbonylé ne possède pas d'atome d'hydrogène sur l'atome de carbone en a du carbonyle. Il n'y a donc pas de réaction concurrente d'énolisation. On s'intéresse ici à l'addition du carbanion dérivé du malonate d'éthyle sue le benzaldéhyde. On utilise souvent une amine comme la pipéridine comme catalyseur. Des acides de Lewis comme ZnCl2 ou SmI sont aussi employés.
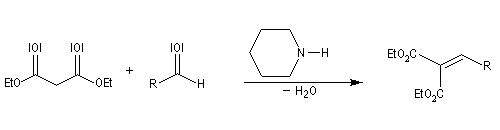
Un mécanisme communément admis est le suivant.
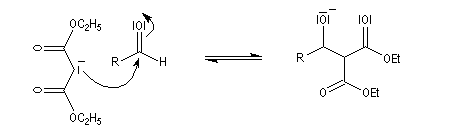
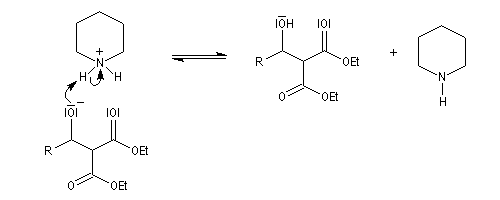
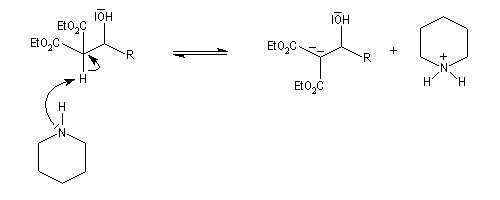
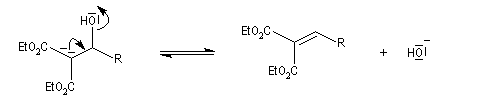
La modification de Doebner de la réaction de Knoevenagel consiste à utiliser la pyridine comme base. Elimination et décarboxylation ont alors lieu en même temps.
Un exemple d'application de la réaction de Knoevenagel à la synthèse de la carbétoxycoumarine est donné à la référence [48].
Condensation de Stobbe
Il s'agit d'une réaction d'addition-élimination entre le carbanion du succinate de diéthyle et un composé carbonylé :
- le succinate de diéthyle est déprotoné par une base forte comme le tertiobutylate de potassium ;
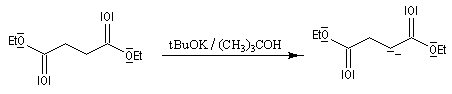
- le carbanion s'additionne sur le composé carbonylé ;
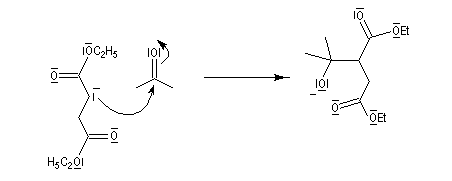
- l'adduit formé peut subir une cyclisation en lactone car il possède un ion éthanolate libérable ;
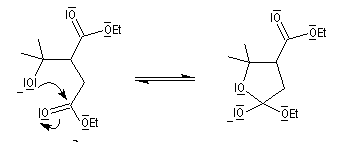
- l'ouverture de la lactone en milieu acide fournit le produit final.
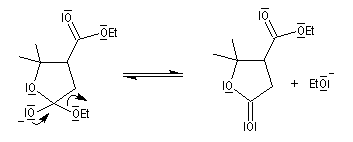
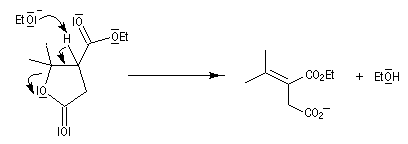
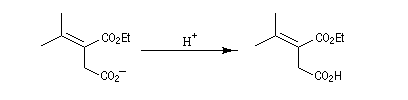
Pour un exemple de cette réaction, voir [42] et [43].
Réaction de Reformatsky
Cette réaction, mise au point par S. Reformatsky en 1887 consiste à faire réagir un bromoester en présence de zinc avec un aldéhyde ou une cétone. Il se forme un organozincique de façon intermédiaire. Celui-ci est inactif vis à vis de la
fonction ester. En revanche, il réagit avec le composé carbonylé plus réactif que l'ester.
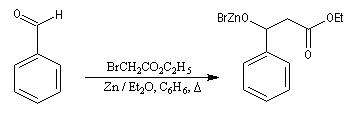
Après hydrolyse du produit d'addition, on obtient un b-hydroxyester.
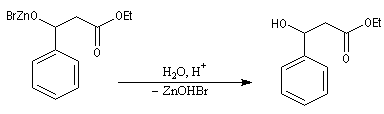
Pour un exemple de réalisation expérimentale, voir [37]. Les b-hydroxyesters sont facilement déshydratés en milieu acide pour donner des esters a, b-éthyléniques. Ceux-ci sont des substrats de départ utiles dans de nombreuses synthèses organiques. Voici un exemple de cette suite de réactions.
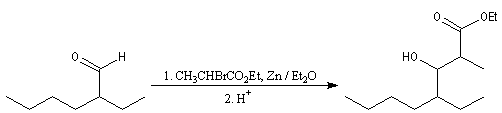
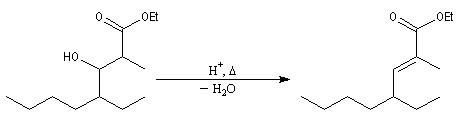
La réaction ci-dessous est une étape de la synthèse de l'équilénine (Bachmann, 1939 [52]). Notons que SOCl2 joue ici le rôle de déshydratant.
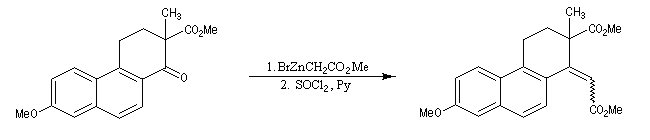
La réaction de Reformatsky peut être rendue