|
|
|
|
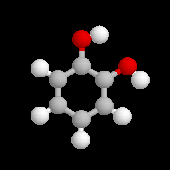
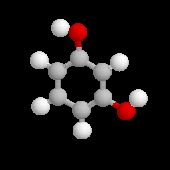
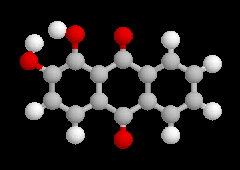
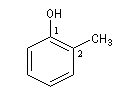
2-méthylphénol |
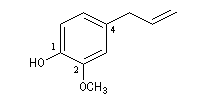
2-méthoxy-4-(prop-2-ényle)-phénol |
Définitions
On appelle phénols les dérivés hydroxylés du benzène et des hydrocarbures aromatiques, dans lesquels le groupe OH est lié à un atome de carbone du cycle benzénique. Les dérivés polyhydroxylés sont appelés polyphénols. Rappelons que chez les alcools le groupe OH est lié à un atome de carbone saturé.
Nomenclature
Ils sont nommés comme des phénols substitués. Le numéro le plus petit est affecté à l’atome de carbone porteur du groupe OH.
|
|
|
|
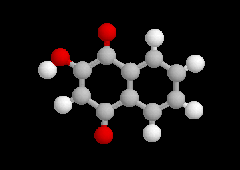
2-méthylphénol |
2-méthoxy-4-(prop-2-ényle)-phénol |
Certains dérivés sont connus sous un nom d'usage. Le 6-isopropyl-2-méthyphénol est le thymol. Le 2-méthoxy-4-(prop-2-ényle)-phénol est l'eugénol. Les phénols dérivés du toluène sont appelés crésols.
Quelques phénols et dérivés importants
|
|
Le phénol est un produit de synthèse. Pur, il se présente à la température ordinaire comme un solide blanc cristallisé. C'est un composé toxique (VME = 19 mg.m-3) qui provoque des brûlures graves sur la peau. Il doit être manipulé en utilisant des gants et des lunettes de protection. Ses solutions ont été parmi les premiers antiseptiques utilisés en médecine (Lister 1867). On l'utilise dans l'industrie comme réactif de base dans la synthèse du cyclohexanol dont la coupure oxydante conduit à l'acide adipique, qui sert à la préparation du nylon 6,6.
|
A l'heure actuelle le phénol est préparé par le procédé Hock qui consiste à oxyder de l'isopropylbenzène (cumène) par le dioxygène de l'air. Le sous-produit de la réaction est la propanone (acétone) qui est également un produit important utilisé notamment comme solvant. Ce procédé particulièrement avantageux illustre une des caractéristiques de la chimie industrielle moderne : limiter le coût des réactifs (ici O2 de l'air) et valoriser au maximum les sous-produits.
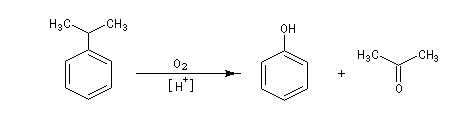
La réaction est de type radicalaire. Le radical benzilique formé possède une certaine stabilité. Il se forme intermédiairement un hydroperoxyde de cumène qui est ensuite décomposé grâce à une catalyse acide. Un procédé plus ancien consistait à effectuer la fusion alcaline d’un sel d’acide sulfonique.
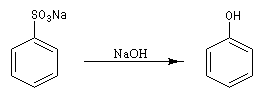
Les substitutions nucléophiles sur un cycle aromatique non désactivé sont toujours difficiles, la réaction nécessite une température élevée (250 °C < T < 300 °C et base forte très concentrée).
Cette réaction peut permettre l'introduction d'un groupe OH sur un cycle aromatique mais en raison des conditions drastiques qu'elle implique, elle est peu utilisée dans la synthèse des molécules complexes et fragiles.
Il est peu vraisemblable que le mécanisme soit du type addition-élimination via un complexe de type Meisenheimer comme chez les aromatiques possèdant des cycles très désactivés. Le mécanisme exact de cette réaction n'est pas connu de façon exacte à l'heure actuelle.
|
|
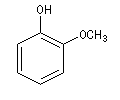
Le gaïacol ou 2-méthoxybenzène est présent comme d'autres phénols dans la fumée issue de la combustion de certaines espèces végétales dont le bois de gaïac (photographie ci-contre).
|
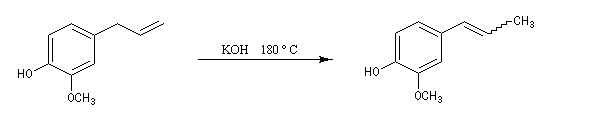
L'eugénol peut être isomérisé en isoeugénol sous l'action de plusieurs réactifs. La force motrice de la réaction est la formation d'une double liaison conjuguée avec un cycle aromatique. On obtient un mélange des deux diastéréo-isomères Z et E
.
|
|
L'eugénol est présent à l'état naturel dans le clou de girofle auquel il donne son goût et son odeur caractéristique. Il est utilisé comme antiseptique par les chirurgiens dentistes. On peut l'obtenir grâce à la transposition de Claisen d'un éther d'allyle du gaïacol. La coupure oxydante de la chaîne latérale de l'isoeugénol conduit à la vanilline selon une réaction mise au point au siècle dernier par Reimer et Tiemann qui ne possède plus qu'un intérêt historique. |
L'oxydation ménagée de l'éthanoate d'isoeugénol (mélange des deux diastéréo-isomères Z et E) peut être réalisée par les ions permanganate en utilisant un milieu diphasé et une catalyse par transfert de phase [33]. Le catalyseur est un ion ammonium quaternaire (aliquat 336) ou catalyseur de Starcks [34].
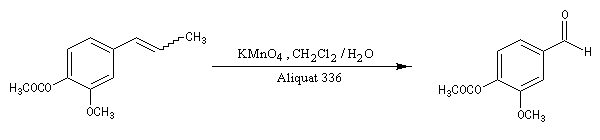
|
|
La vanilline se rencontre à l'état naturel dans la gousse de vanille et le benjoin de Siam. Son parfum et son goût délicats en font le composé le plus utilisé comme arôme. La vanilline a été utilisée comme matière première dans la synthèse de la L-DOPA, le seul médicament dont on dispose à l'heure actuelle contre la maladie de Parkinson. Il est amusant de remarquer que la vanilline d'origine naturelle coûte environ 250 fois plus cher que la même molécule obtenue par synthèse ! |
Structure de la molécule de phénol
L'énergie de résonance évaluée grâce à la réaction d'hydrogénation vaut 167 kJ.mol-1. Elle est donc plus élevée que l'énergie de résonance du benzène (150 kJ.mol-1). On interprète ce résultat par la participation d'un doublet non liant de l’atome d’oxygène à la résonance. Les mesures aux rayons X montrent que la molécule est plane ce qui autorise une délocalisation maximale. Cette participation à la délocalisation électronique se traduit aussi par le raccourcissement de la longueur de la liaison C-O et par l'augmentation de l'énergie de cette liaison par rapport à celle d'un alcool comme le cyclohexanol.
Constantes physiques
Les températures de changement d'état des phénols sont plus élevées que celle des hydrocarbures de même masse molaire. On l'interprète par le fait que ces composés sont associés par liaison hydrogène intermoléculaire. Le phénol lui même est un solide à la température ordinaire.
|
TF / °C |
TE / °C |
s (H2O) /g.L-1 |
m / D |
|
41 |
181 |
93 |
1,59 (Ph vers OH) |
La miscibilité avec l'eau dépend beaucoup de la température. Elle devient totale si T > 63 °C.
Certains composés polyfonctionnels comme le 2-hydroxybenzaldéhyde (salicylaldéhyde) possèdent une liaison hydrogène intramoléculaire (s # 3480 cm-1). Ce type de liaison se distingue facilement d'une liaison intermoléculaire. En effet, un tel pic n'est pas affecté lors de la dilution du composé dans un solvant inerte comme CCl4.
Spectroscopie UV-Visible
Le phénol absorbe dans l'ultraviolet. Ses solutions sont incolores. La déprotonation et le passage à l'ion phénolate provoquent un effet bathochrome (déplacement de la bande d'absorption vers les grandes longueurs d'onde) et hyperchrome (renforcement de l'intensité de l'absorption).
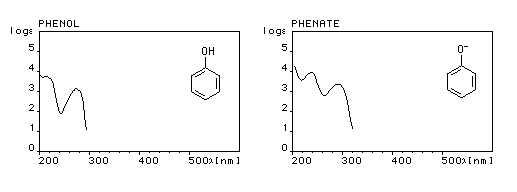
Le nitrophénol possède une bande d'absorption centrée à 270 nm. Il absorbe dans l'ultraviolet et, de ce fait, il est incolore. Par addition de soude il est transformé quantitativement en sa base conjuguée : l'ion nitrophénolate qui absorbe dans le visible lm # 400 nm et qui possède une couleur jaune. De ce fait, le système nitrophénol - ion nitrophénolate est utilisé comme indicateur coloré acido-basique.
L'oxydation du phénol lui même peut avoir lieu sous l'action de très nombreux oxydants : Fe3+, O2, etc. symbolisés par [O]. Elle passe par la formation de radicaux libres phénoxyles relativement stables, qui évoluent pour donner par couplage des produits complexes souvent colorés, dont la structure est mal définie. C'est la raison pour laquelle les récipients contenant du phénol doivent être soigneusement conservés à l'abri de l'air.
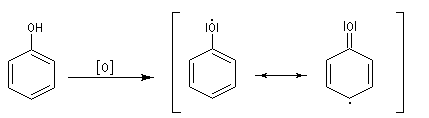
Certains phénols comme l'a-tocophérol peuvent jouer le rôle de capteurs de radicaux libres.
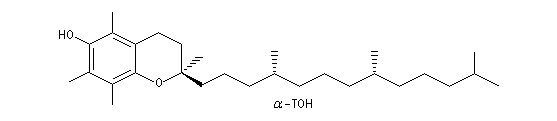
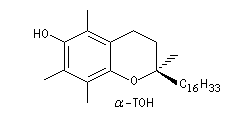
![]()
Acidité
Comparons les pKa des couples phénol/phénolate et cyclohexanol/cyclohexanolate :
|
Couple |
PhOH/PhO- |
CyOH/CyO- |
|
pKa |
9,9 |
18 |
Le phénol est donc environ cent million de fois plus acide que le cyclohexanol.
De ce fait, il est déprotoné de façon quantitative par une solution aqueuse d'ions hydroxyde pKa (H2O/OH-) = 14 pour donner une solution de phénolate de sodium.
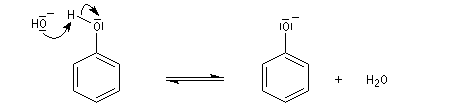
D'une façon générale, les phénols sont beaucoup plus acides que les alcools. La charge négative dispersée dans le cycle, est mieux supportée par la structure et la stabilisation qui en résulte est à l'origine de la diminution de la basicité. On peut rendre compte de cette propriété en écrivant les formes de résonance suivantes :
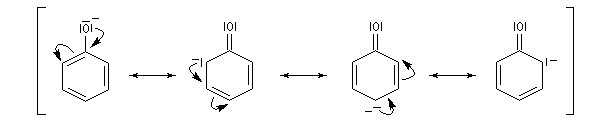
Les charges négatives apparaissent sur les atomes de carbone en position ortho et para. L'acidité est accrue par la présence de groupes attracteurs sur le cycle.
|
|
|
Classiquement, on interprète l'accroissement de stabilité de la base conjuguée par la résonance du doublet non liant de l'oxygène avec le cycle aromatique substitué par les groupes nitro attracteurs inductifs et mésomères.
Puisqu'il s'agit d'acidité relative à un solvant donné en l'occurrence l'eau et non d'acidité en phase gazeuse, il faut faire attention que la solvatation joue ici un rôle très important et il faut tenir compte de la solvatation différente de l'acide et de sa base conjuguée.
Basicité
Les phénols sont des bases beaucoup plus faibles que les alcools : pKa (PhO+H2/PhOH) = - 7
On peut l'interpréter par une protonation de l'oxygène beaucoup plus difficile que chez les alcools du fait de la délocalisation du doublet.
Propriétés nucléophiles de l'atome d'oxygène
Formation de complexes
Les ions phénolate peuvent jouer le rôle de ligands par leur atome d'oxygène négatif.
|
|
Les ions phénolate donnent des complexes de transfert de charge intensément colorés avec certains cations de métaux de transition tels que les ions Fe3+.
|
La réaction peut servir de test des phénols car la valeur de la longueur d'onde lm d'absorption dépend des substituants portés par le cycle. Les groupes donneurs (alkyle, hydroxy, etc.) provoquent un effet bathochrome (déplacement de l'absorption vers des longueurs d'onde plus grandes). Il faut cependant faire attention que d'autres composés comme les acides carboxyliques donnent des complexes colorés avec l'ion Fe3+. Ce test n'est donc pas caractéristique des phénols.
Voir aussi les complexes métalliques avec le ligand salen.
Alkylation : synthèse d'éthers
Les ions phénolate sont de meilleurs nucléophiles que les ions alcoolate bien que de basicité plus faible car la présence du cycle aromatique les rend très polarisables. L'alkylation des ions phénolate sur l'oxygène est un cas particulier de la synthèse de Williamson des éthers.
Avec un halogénure d'alkyle primaire, le mécanisme est de type SN2 et dans ce cas, la réaction est accélérée dans un solvant dipolaire aprotique (DMSO, DMF) qui solvate le contre-ion et exalte le caractère nucléophile de l'oxygène.
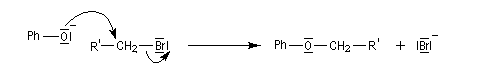
Avec un halogénure tertiaire, le mécanisme est de type SN1 car le substrat forme facilement un carbocation.
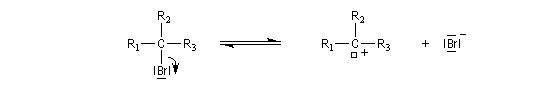

Les ions phénolate sont plus nucléophiles que les ions alcoolate ainsi que l'illustre la synthèse de la codéine à partir de la morphine.
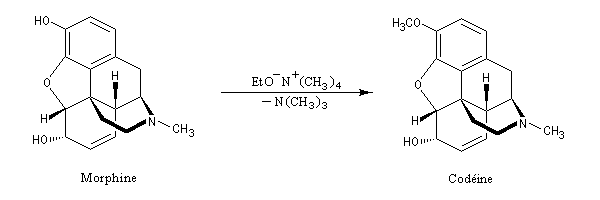
Acylation : synthèse d'esters
La diminution du caractère nucléophile de l'oxygène, quand on passe des alcools aux phénols, apparaît nettement dans la réaction d'acylation. Les phénols sont acylés par les halogénures d’acyle et par les anhydrides mais pas par les acides carboxyliques. Rappelons qu'avec les alcools, cette réaction forme un ester par une transformation renversable.
Le mécanisme de la réaction implique une addition nucléophile de l'oxygène du phénol sur le groupe carbonyle du chlorure d'acyle, suivie d'une fragmentation de l'intermédiaire tétraédrique formé.
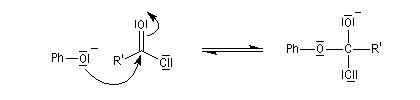
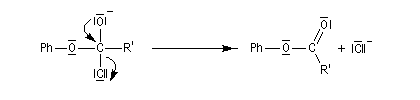
Il est donc du même type que le mécanisme d'acylation des alcools.
Le dérivé acétylé de l'acide salicylique est l'acide 2-acétyloxybenzoïque couramment appelé acide acétylsalicylique et qui est commercialisé sous le nom d’aspirine.
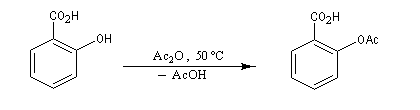
|
|
Le strasbourgeois Gerhardt a préparé en 1853 le dérivé acétylé de l'acide salicylique que Félix Hoffmann utilisa comme médicament pour soulager les rhumatismes de son père. La société Bayer a commercialisé le produit en 1899 sous le nom d'aspirine (A pour acétyl, spir pour spirée et ine terminaison générique pour les substances issues du règne végétal. |
|
|
Dès le IVe siècle avant J.C, Hippocrate prescrivait des décoctions d'écorce de saule (image de gauche) pour combattre les fièvres. La salicyline est un composé naturel présent dans l'écorce de Saule. C'est un hétéroside du D-glucose.
|
Réactions de substitution affectant le cycle
Activation et régiosélectivité
Le groupement OH est un activant fort et les réactions de substitutions électrophiles sont donc beaucoup plus rapides qu'avec le benzène. Un catalyseur n'est généralement pas nécessaire. On doit utiliser des conditions expérimentales particulières lorsque l'on veut éviter les polysubstitutions.
Le groupe OH oriente exclusivement en ortho et para.
Bromation
Elle est réalisable sans catalyseur par réaction entre le dibrome et l'ion phénolate.
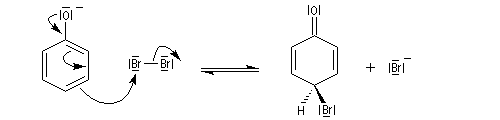
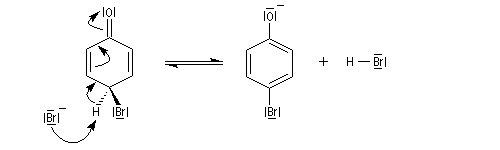
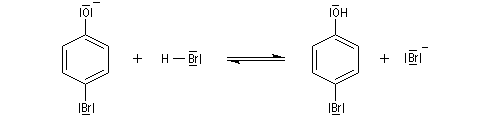
A la température ordinaire, en milieu aqueux, la réaction ne s'arrête pas à la monobromation. Elle se poursuit sur les positions ortho et on obtient finalement le tribromophénol. Cette réaction témoigne de la grande réactivité du phénol si on la compare à celle du benzène. Rappelons qu'avec le benzène un acide de Lewis comme AlCl3 est nécessaire et à froid, la réaction s'arrête au stade de la monobromation.
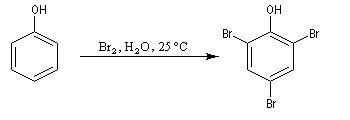
Le tribromure de tétrabutylammonium permet de s'arrêter à la monobromation en position para [21].
|
|
Le bécher contient une solution d'eau de brome dans laquelle on verse progressivement une solution aqueuse de phénol. On obtient immédiatement un abondant précipité blanc de 2,4,6-tribromophénol. La réaction peut servir à effectuer le dosage du phénol en utilisant une solution d'eau de brome de concentration connue en excès et une solution titrée d'iodure de potassium. Le dibrome en excès oxyde I-en I2 et le diiode peut être dosé en retour avec une solution titrée de thiosulfate. |
|
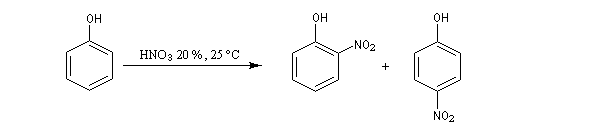
Composé |
2-nitrophénol (ortho) |
4-nitrophénol (para) |
|
% |
35 |
15 |
En milieu acide dilué l'entité électrophile est l'ion NO+. Ecrivons le mécanisme pour la formation du dérivé para.
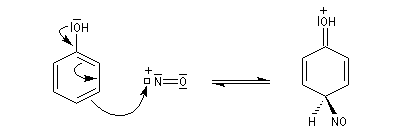
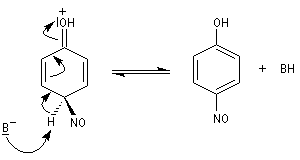
Le nitrosophénol est oxydé en nitrophénol.
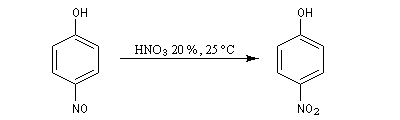
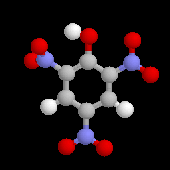
Notons que les nitrophénols sont des composé particulièrement toxiques.
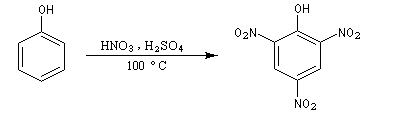
|
|
L'acide picrique, découvert par Woulfe en 1771 a été préparé par J. M Haussmann en 1788 par réaction entre l'acide nitrique et l'indigo. C'est le premier colorant synthétique. L'ion picrate forme avec les ammonium terminaux de la laine, de la soie, du nylon, une liaison ionique.
|
Carboxylation
L'introduction directe d'un groupe acide carboxylique dans le cycle aromatique d'un phénol est réalisable alors qu'elle ne l'est pas pour le benzène.
La réaction de Kolbe-Schmidt consiste à faire passer un courant de CO2 gazeux dans du phénolate de sodium solide, fluidisé, à température et pression élevées (180 °C, 100 bar).
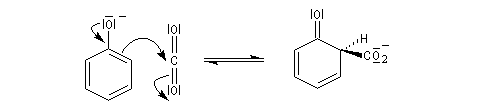
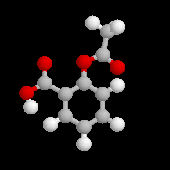
Le produit de la réaction est le 2-hydroxybenzoate de sodium (salicylate de sodium).
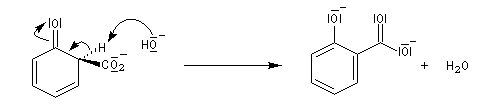
L’acylation de l’acide conjugué, l'acide 2-hydroxybenzoïque ou acide salicylique conduit à l’aspirine.
Synthèse de la phénolphtaléine
La réaction entre le phénol et l'anhydride phtalique en présence de ZnCl2 comme catalyseur permet la synthèse d'un colorant utilisé comme indicateur acido-basique : la phénolphtaléine.
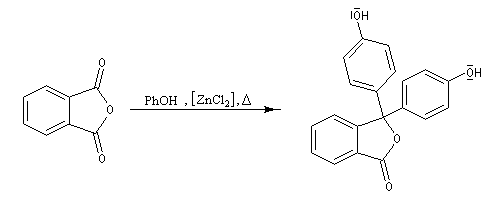
Pour pH < 7, la phénolphtaléine est un composé moléculaire noté PH qui, comme le phénol, absorbe dans l'U.V et qui est incolore. Pour pH > 10, une fonction phénol est déprotonée. L'intermédiaire initialement formé est instable et conduit à l'ion P-dans lequel l'atome de carbone joignant les cycles est dans un environnement localement plan ce qui permet une délocalisation des électrons p sur un système assez vaste pour que le maximum de la bande d'absorption soit située dans le visible (lm = 554 nm). La solution est fortement colorée en rose.
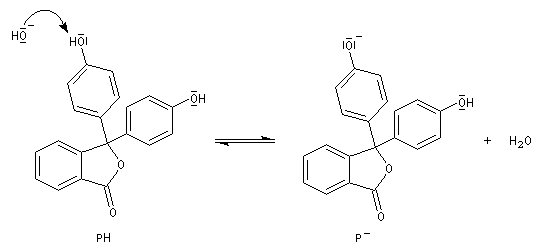

Si le mélange est rendu très basique par addition d'ions hydroxyde en solution concentrée, la solution se décolore lentement. Les ions OH- s'additionnent sur l'atome de carbone électrophile insaturé de la structure quinonique pour donner l'ion POH2-. La structure conjuguée quinonique a disparu. La conjugaison est beaucoup moins étendue et le composé est incolore.

|
|
La phénolphtaléine est un indicateur coloré très largement utilisé dans les dosages acido-basiques. Sa zone de virage (de l'incolore au rose) étant comprise entre 8,2 et 10 on l'utilise lorsque le pH à l'équivalence est voisin de 9. A titre d'exemple, le repérage de l'équivalence du dosage de l'acide éthanoïque pKa (AcOH/AcO- ) = 4,7 de concentration 0,1 mol.L-1 par une solution de soude de concentration 0,1 mol.L-1 peut être détecté par le changement de couleur de cet indicateur. |
Hydroxyméthylation
Le phénol réagit avec le méthanal dans la réaction d'hydroxyméthylation. Celle-ci rappelle la condensation aldol mais ici l'équilibre de tautomérie est en faveur de la forme phénol qui est la plus stable en raison de son caractère aromatique.
En milieu acide le carbonyle est protoné. Il y a une activation électrophile.
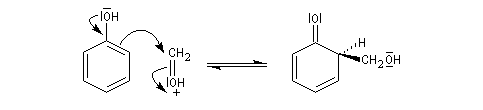
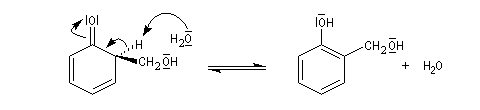
Le composé obtenu dans la réaction d'hydroxyméthylation peut perdre de l'eau par chauffage. On obtient un intermédiaire appelé quinométhane. La réaction qui implique les positions ortho et para est catalysée par les ions H+ ou OH-. Ecrivons la réaction en milieu acide.
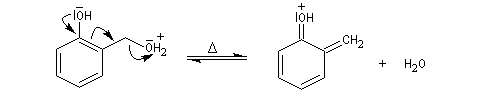
Le composé précédent réagit facilement avec un excès de phénol selon une réaction de type Michaël (addition conjuguée sur un composé carbonylé a,b-éthylénique
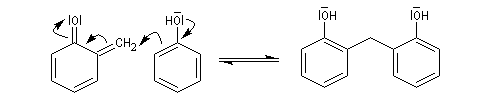
Et ainsi de suite. Les positions ortho et para peuvent être toutes impliquées par polymérisation. On obtient finalement une résine thermodurcissable, de structure tridimensionnelle : la bakélite. On a représenté ci-dessous une structure simplifiée bidimensionnelle.
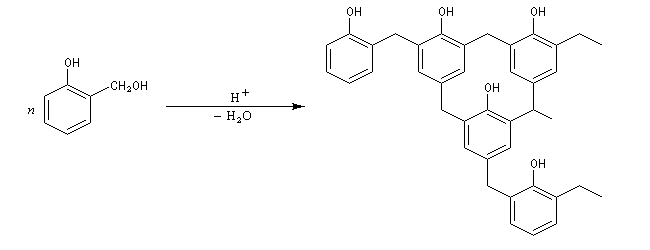
La bakélite fut l'un des premiers polymères industriel. Ce composé a été préparé par le chimiste américain d'origine Belge L.H Bakeland (1863-1944) qui fonda une usine de production (Bakelite corp). La bakélite possède des usages nombreux et variés (contre-plaqués, plastiques moulés, revêtements de têtes de fusées).
Alkylation des halogénures allyliques
Les ions phénolate peuvent être alkylés au niveau de l'atome d'oxygène (O-alkylation), dans des solvants aprotiques comme le DMF, pour conduire aux éthers. Ils peuvent aussi réagir au niveau des atomes de carbone en position ortho et para (C-alkylation). Comme les ions énolate, ce sont des nucléophiles ambidents.
Les halogénures d'allyle donnent assez facilement des carbocations allyliques stabilisés par résonance.
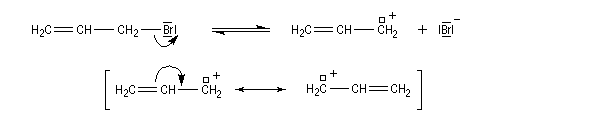
L'alkylation du phénolate sur l'oxygène (O-alkylation) conduit au prop-2-ényloxybenzène (oxyde d'allyle et de phényle) par la réaction de Williamson.

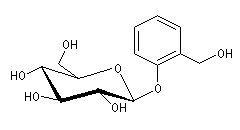
Le produit correspondant à la réaction sur la position ortho est le 2-(prop-2-ényl)-phénol (orthoallylphénol.)
La C-alkylation est favorisée dans les solvants protiques qui, comme l'eau, contractent des liaisons hydrogène et solvatent l'atome d'oxygène. La C-alkylation est cependant moins favorisée que la O-alkylation car l'aromaticité du cycle est perdue au cours de la réaction.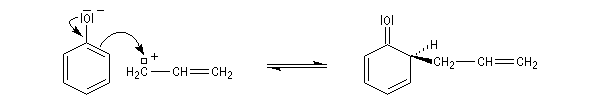
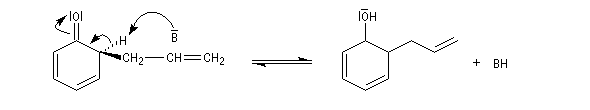
Les pourcentages des produits précédents dépendent des conditions expérimentales : température, nature du solvant, type de contre-ion.
Lorsque la réaction est conduite dans les conditions d'un contrôle cinétique (température basse, durée courte) le produit de O-alkylation est majoritaire. Sous contrôle thermodynamique (équilibre entre les composés) c'est le produit de C-alkylation qui prédomine. Le produit de O-alkylation se forme plus rapidement que celui de C-alkylation car dans le second cas l'état de transition perd son caractère aromatique. Les éthers d'allyle et de phényle subissent la réaction de transposition de Claisen.
Réaction de Reimer-Tiemann
Les aldéhydes aromatiques peuvent être obtenus par formylation directe des hydrocarbures aromatiques au moyen d'un mélange de CO, HCl et d'un acide de Lewis tel que AlCl3 jouant le rôle de
catalyseur. Cette réaction de Gatterman-Koch n'est pas facilement utilisable avec les phénols, ni avec les éhers après protection de la fonction, car ces derniers complexent l'acide de Lewis et bloquent son activité catalytique.
La réaction de Reimer-Tiemann permet la formylation des phénols. L'espèce réactive est un dichlorocarbène formé in-situ par réaction entre le chloroforme et les ions hydroxyde. On obtient majoritairement le composé ortho et dans une proportion moindre le para. Cette réaction a constitué la première observation d'un carbène (J. Hine 1950) [32].
La formation du dichlorocarbène est exemple d'a-élimination car l'hydrogène arraché et le nucléofuge sont liés au même atome de carbone.
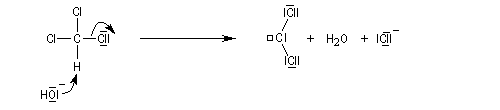
Le dichlorocarbène est très réactif. La première étape est l'addition de cet intermédiaire sur le cycle aromatique.
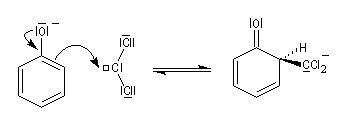
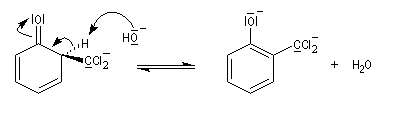
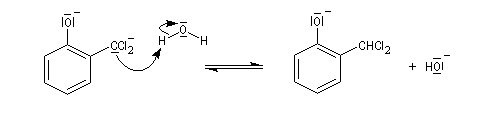
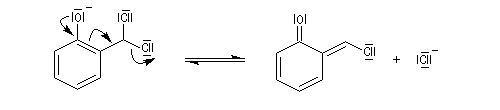
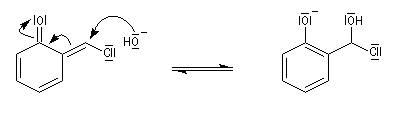
prototropie.
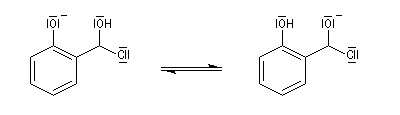
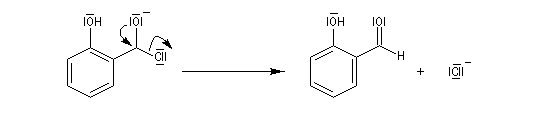
Au cours de la réaction, il y a formation des isomères ortho et para. L'isomère ortho possède une liaison hydrogène intramoléculaire. Il est insoluble dans l'eau. Les deux isomères peuvent être facilement séparés par entraînement à la vapeur.
L'aldéhyde salicylique permet la préparation du ligand salen.
Remarque : autre exemple de formylation, sur les amines aromatiques cette fois : la réaction de Vilsmeier-Haack.|
|
Le catéchol (pyrocatéchol) est le 1,2-dihydroxybenzène. Il peut être obtenu à partir de l'orthochlorophénol par la réaction de fusion alcaline. On l'utilise comme antioxygène car il inhibe les réactions en chaîne d'oxydation en captant les radicaux. De même, il empêche la polymérisation spontanée de certains composés éthyléniques comme le styrène. On l'élimine de ce dernier en ajoutant de la soude au mélange. Le catéchol est déprotoné en milieu basique et les ions passent en phase aqueuse. On sépare le styrène et la phase aqueuse par décantation. |
|
|
Le résorcinol est le 1,3-dihydroxybenzène. Son oxydation est beaucoup plus difficile que celle de ses deux isomères, le catéchol et l'hydroquinone car il n'existe pas de quinone correspondante. L'oxydation du résorcinol conduit à CO2 et H2O. Sa réaction avec l'anhydride phtalique fournit la fluorescéine qui est un colorant. |
|
|
|
Le poison ivy
Le 3-Pentadécylcatechol est un dérivé alkylé du catéchol. Ce composé, représenté ci-dessous est l'un des constituants principaux d'une substance irritante synthétisée par une plante commune en Amérique du Nord : le poison Ivy.
|
|
|
|
|
L’alizarine est un composé de couleur rouge synthétisé par W.H. Perkin en 1868 (qui s'était déjà distingué en préparant la mauvéine à l'âge de 18 ans). Ce fut le premier colorant d'origine naturelle obtenu par synthèse. Sa production industrielle entraîna la ruine des cultivateurs de garance du sud de la France à partir de laquelle on préparait le composé avant la mise au point de sa synthèse. Pour tenter d'enrayer la chute des cours, le gouvernement de l'époque décida d'utiliser les surplus de matière colorante pour teindre les pantalons des soldats d'infanterie ! C'est dans cet uniforme que les soldats français montèrent au front au début de la guerre de 1870. On peut se faire une idée de cet uniforme, sur la célèbre toile d' E. Manet : le joueur de fifre. |
|
|
|
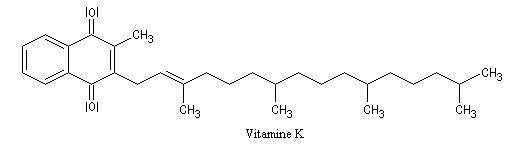
Vitamine K1
La vitamine K1 est une naphtoquinone qui possède des propriétés anti-hémorragiques.
Réactions d'oxydo-réduction des diphénols et des quinones
L'oxydation ménagée du benzène-1,2-diol ou hydroquinone est réalisable avec différents oxydants (FeCl3, Ag2O, Na2Cr2O7). Elle conduit à la cyclohexa-2,5-diène-1,4-dione ou parabenzoquinone.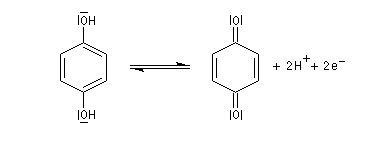
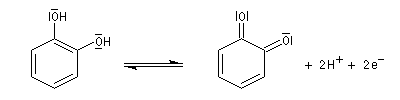
L'oxydation du catéchol fournit la quinone correspondante mais avec un moins bon rendement.
|
|
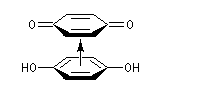
Hydroquinone et benzoquinone forment un complexe [1,1] appelé quinhydrone. Il s'agit d'un complexe de transfert de charge dans lequel l'hydroquinone joue le rôle de donneur et la benzoquinone celui d'accepteur. A la température ordinaire, la quinhydrone se présente sous la forme de cristaux intensément colorés en vert (TF = 171 °C) peu solubles dans l'eau. Une solution aqueuse de quinhydrone se comporte comme un mélange équimolaire de quinone Q et d'hydroquinone H2Q. La solution hydroalcoolique contenue dans le bécher possède une couleur jaune que lui confère la benzoquinone. |
La quinhydrone est un complexe acide-base de Lewis de type p
Le système quinone Q-hydroquinone H2Q présente l'importante propriété pour un système organique d'être réversible :
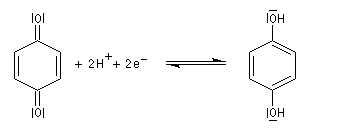
Le potentiel standard du couple vaut (298 K) : Eo = 0,7 V
Le potentiel d'un fil de platine plongeant dans une solution aqueuse de quinhydrone est donné par la formule de Nernst. Puisque [Q] = [H2Q], il a pour expression (T = 298 K) :
E = Eo - 0,06 pH
Ce système a été utilisé comme électrode de pH avant que ne se généralise l'emploi des électrodes de verre. La linéarité entre le pH et le potentiel n'est rigoureuse qu'en deça de pH = 9 car, au dela, l'hydroquinone est déprotonée pour donner sa base conjuguée QH- .
Les propriétés réductrices de l'hydroquinone expliquent son utilisation comme révélateur en photographie. La réduction s'effectue en milieu basique (pH = 12) et ne concerne que les ions Ag+ qui ont été activés par la lumière.
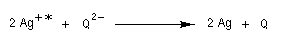
Oxydation en milieu biologique
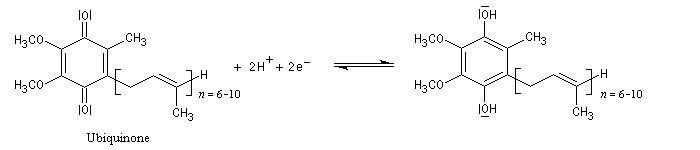
Le système Q/QH2 possède un potentiel ("standard apparent" dans les conditions biologiques) E'o= 0,10 V. L'ubiquinone intervient dans la chaîne respiratoire en tant que transporteur d'équivalents réducteurs. Au cours d'une cascade de réactions, l'ubiquinone Q est réduite en QH2 qui est à son tour réoxydée en Q.
![]()
![]()
|
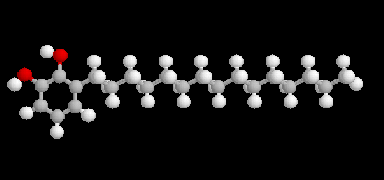
L'ubiquinone ou coenzyme Q comporte une structure de type quinone sur laquelle est greffée une chaîne isoprénique de longueur variable
comportant entre 6 et 10 unités isoprène. L'ubiquinone se rencontre dans le règne animal et dans le règne végétal. C'est à son don d'ubiquité que cette
molécule doit son nom. La molécule représentée ci-dessus est telle que n = 6.
La chaîne latérale lipophile permet à la molécule un ancrage dans
la membrane interne des mitochondries. |
Réactions d'addition
Additions conjuguées sur les quinones
En milieu acide, le carbonyle est protoné c'est un exemple d'activation électrophile..
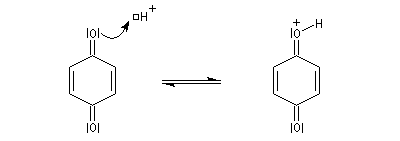
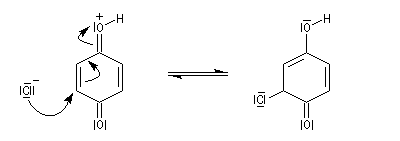
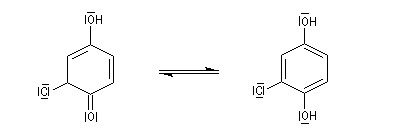
La réaction avec un acide benzènesulfonique conduit à un composé facilement cristallisable.
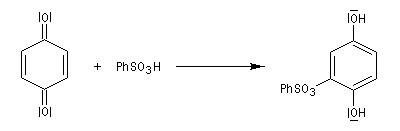
Réactions de Diels-Alder des quinones
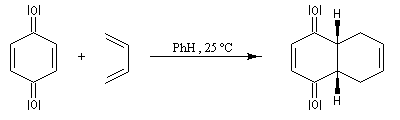
En milieu acide, la dicétone se tautomérise en diphénol beaucoup plus stable du fait du cycle aromatique.
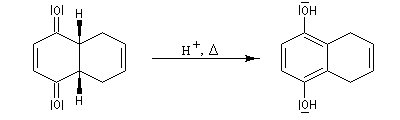
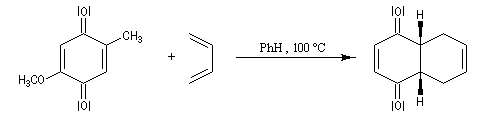
Bibliographie
[1] Manuel d'expériences de chimie - UNESCO Société chimique de France - Université de Montpellier.
[2] P. Rendle, M. V. Vokins, P. Davis, Experimental Chemistry (Edward Arnold).
[3] L. F. Fieser, K. L. Williamson - Organic Experiments (D. C. Heath and Company).
[4] R. Adams, J. R. Johnson, C. F. Wilcox - Laboratory Experiments in Organic Chemistry (The Macmillan Company, Collier-Macmillan Limited).
[5] G. K. Helmkamp, H. W. Johnson Jr, Selected Experiments in Organic Chemistry (W. H. Freeman and Co).
[6] Vogel's Textbook of Practical Organic Chemistry (Longman).
[7] J. R. Mohrig, D. C Neckers, Laboratory Experiments in Organic Chemistry (D. Van Nostrand Company).
[8] R. Q. Brewster, C. A. Van der Werf, W. E. Mc Ewen, Unitized Experiments in Organic Chemistry (D. Van Nostrand Company).
[9] F. G. Mann, B. C. Saunders, Practical Organic Chemistry (Longman).
[10] M. T. Yip, D. R. Dalton, Organic Chemistry in the Laboratory (D. Van Nostrand Company).
[11] Journal of Chemical Education.
[12] S. Hünig, E. Lücke, W. Brenninger, Organic Syntheses.
[31] Woodward, R. B. ; Sondheimer, F. ; Taub, D. ; Heusler K. ; McLamore W. M. J. Am. Chem. Soc. 74, 4223 (1952).
[32] Hine J, J. Am. Chem. Soc. 1950, 72, 2438.
[33] Lampman G. M, Sharpe S. D, A Phase Transfer Catalyzed Permanganate Oxydation. Preparation of Vanilin from Isoeugenol Acetate - J. Chem. Ed., 60, 1983, p. 503-504.
[34] C.M. Starks (1971). "Phase-transfer catalysis. I. Heterogeneous reactions involving anion transfer by quaternary ammonium and phosphonium salts". J. Amer. Chem.93 : 195
[35] Reimer, K.; Tiemann, F. Ber. 1876, 9, 824 & 1268 & 1285.
[14] l'aspirine histoire de l'aspirine
[15] poison ivy compléments sur le poison ivy
[16] The Reimer Tiemann reaction by H. Wynberg and E. W. Meijer
[17] 2,6-dibromo-4-nitrophenol by W. Hartman and J. B. Dickey
[18] Preparation du 2-nitrophenol
[20] 2,6-dibromo,4-nitrophenol
[21] Bromation régiosélective en série aromatique
[22] composants présents dans le Whisky
A. Streitwieser Jr, C. H. Heathcock - Introduction to Organic Chemistry, Macmillan Publishing Co.
J. March - Advanced organic chemistry, Wiley Interscience.
F. A. Carey, R. S. Sunberg - Advanced Organic Chemistry, Plenum Press 1990.
J. Koolman K-H. Röhm - Atlas de biochimie (Médecine- Sciences Flammarion).
N. T. Ahn - Introduction à la chimie moléculaire, Ellipses 1994.
R. Brückner - Mécanismes réactionnels en chimie organique, De Boeck Université 1999.
J. C. Chottard, J. C. Depezay, J. P Leroux - Chimie fondamentale, Hermann 1982.
P. Laszlo - Logique de la synthèse organique. Cours de l'Ecole Polytechnique, Ellipses, 1993.
Vous pouvez, si vous le souhaitez, utiliser le contenu de cette page dans un but pédagogique et non commercial.
Texte, dessins, photographies : Gérard Dupuis - Lycée Faidherbe de LILLE
mai 2014
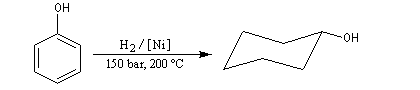

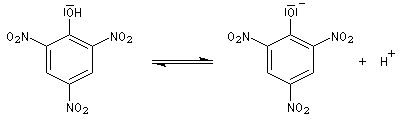

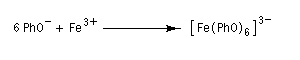

 .
.