Cours de chimie Organique - G. Dupuis - Lycée Faidherbe de Lille
Eléments de stéréochimie dynamique
Définitions
La stéréochimie dynamique s'intéresse aux relations entre le déroulement des réactions chimiques
et la stéréochimie des composés impliqués dans ces réactions. L'analyse conformationnelle fait partie intégrante de la stéréochimie dynamique.
Conformations
Série acyclique
Le citral et le b-cyclocitral sont des isomères de fonction. L'isomérisation du citral C en b-cyclocitral D est catalysée par les ions H+.
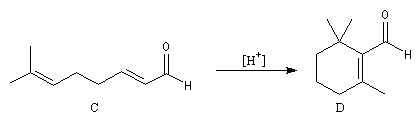
Cet exemple illustre l'importance qu'il y a dans certains cas à prendre en considération certaines conformations particulières d'une molécule.
Dans l'étape de cyclisation, la molécule doit adopter une conformation permettant le rapprochement à une distance convenable des extrémités de la molécule.
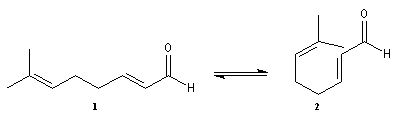
La conformation (2) est énergétiquement défavorisée par rapport à (1) à cause de la présence des nombreuses éclipses entre les liaisons. Bien que très minoritaire par rapport à (1), la consommation de (2) dans les étapes ultérieures déplace l'équilibre conformationnel.
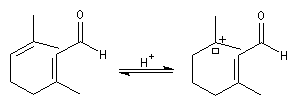
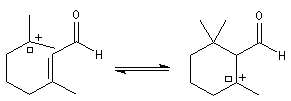
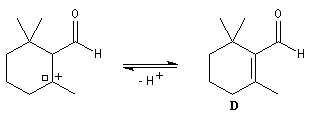
La force motrice de la réaction est la formation de l'a, énone D.
Série cyclique
Les effets conformationnels en série cyclohexanique peuvent être étudiés en utilisant des cycles substitués par un groupement volumineux comme le groupe tertiobutyle. Les dérivés substitués de la (trans)-décaline ont été aussi utilisés.
Les résultats ci-dessous concernent les vitesses relatives d'acétylation par l'anhydride acétique des diastéréoisomères cis et trans des tertiobutylcyclohexanols selon E. L. Eliel [1].
|
Composé |
R2 (cis) |
R1 (trans) |
|
Vitesse relative d'acétylation |
1 |
3,7 |
Ils peuvent être interprétés grâce au mécanisme de la réaction :
- l'étape cinétiquement déterminante est l'attaque nucléophile du groupe hydroxyle de l'alcool sur le carbonyle de l'anhydride qui conduit à un intermédiaire tétraédrique ;
- la fragmentation de l'intermédiaire tétraédrique intervient dans une seconde étape plus rapide.
La différence d'enthalpie libre standard entre les tertiobutylcyclohexanols cis et trans, peut être évaluée à partir des préférences équatoriales des substituants qui sont données dans les tables. Elle est voisine de 3 kJ.mol-1. Pour les intermédiaires I1 et I2, on peut s'attendre à une différence beaucoup plus élevée du fait de substituants plus volumineux. Le diagramme énergétique suivant résume la situation.
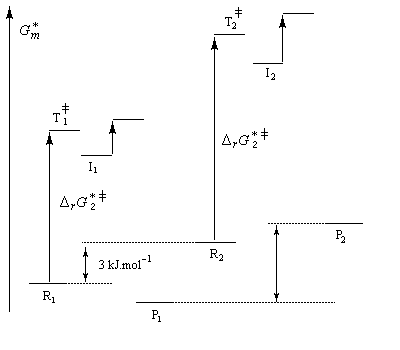
|
Intermédiaire |
I2 |
I1 |
|
Les schémas ci-contre représentent les intermédiaires I1 et I2 à l'aide des
rayons de Van der Waals des atomes (représentation spacefill). On met ainsi en évidence les interactions 1, 3-diaxiales dans I2 qui n'apparaissent pas dans I1. |
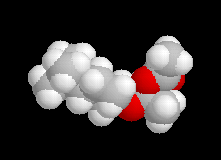
|
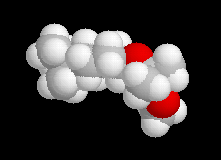
|
Ces intermédiaires sont représentés ci-dessous en utilisant la projection de Newman.
|
Intermédiaire |
I2 |
I1 |
|
Structure |
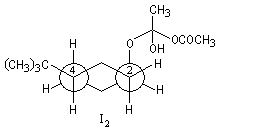
|
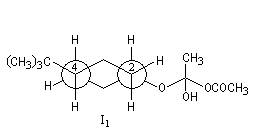
|
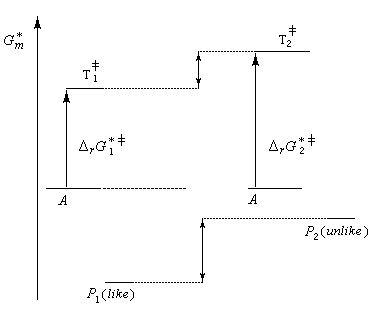
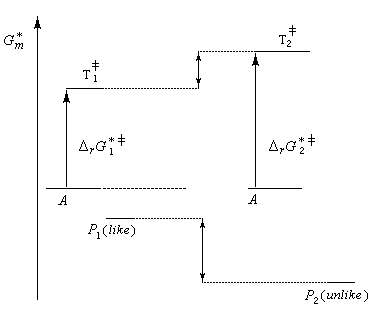
Principe de Curtin-Hammett
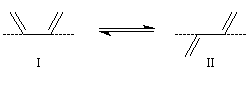
On s'intéresse au cas où des réactifs notés R1 et R2 en équilibre rapide, sont l'objet de réactions compétitives. R1 conduit au produit P1 tandis que R2 conduit à P2.
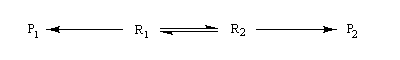
Cette situation se rencontre notamment lorsque deux conformères réagissent avec des vitesses différentes. La plupart du temps, en effet, le passage d'une conformation à l'autre ne nécessite qu'une faible réorganisation de la structure. L'enthalpie libre d'activation correspondante est donc petite en comparaison de celles qui sont mises en jeu dans les réactions des conformères vis à vis du réactif.
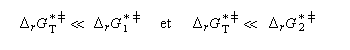
Dans ces conditions, la vitesse d'interconversion entre les conformères est beaucoup plus rapide que celle des réactions compétitives dans lesquelles ils sont engagés.
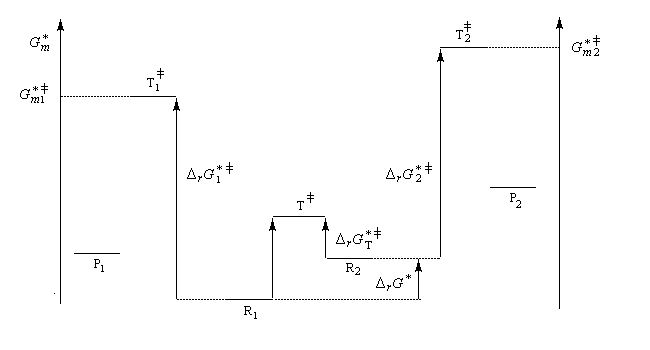
Le diagramme ci-dessous illustre la situation.
Nous allons faire l'hypothèse que la proportion des produits P1 et P2 est le fruit d'un contrôle cinétique. Dans ces conditions, le rapport des quantités de produits formés dépend de leur vitesse de formation :
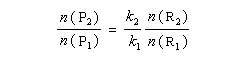
Les réactifs sont en équilibre rapide avec une constante thermodynamique K*.
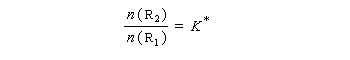
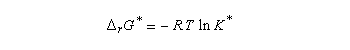
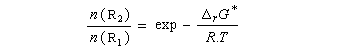
Utilisons la formule d'Eyring afain de relier les constantes de vitesses aux enthalpies libres standard d'activation.
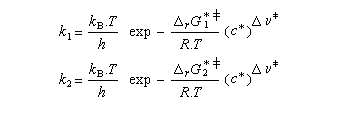
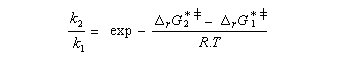
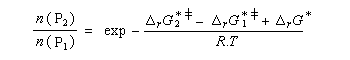
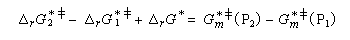
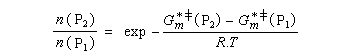
On en conclut que la proportion des produits obtenus dépend de la différence des enthalpies libre d'activation de référence des états de transition conduisant à P1et P2 et non de l'enthalpie libre d'activation pour le passage d'un conformère à l'autre. Ce résultat est connu sous le nom de principe de Curtin-Hammett.
Lorsque les réactifs sont des conformères, c'est la conformation la plus réactive qui intervient pour déterminer la quantité de produit final même si celle-ci est la moins abondante à l'équilibre.
Comme exemple d'application du principe de Curtin-Hammett, considérons la réaction de Diels-Alder entre un diénophile comme le dicyano-1,2-éthène et le buta-1,3-diène.
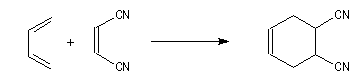
Dans cette réaction, le diène réagit dans la conformation s-cis (I) bien que celle-ci soit moins stable que
la conformation s-trans (II) car cette dernière conduit à l'état de transition d'enthalpie libre minimale.
Naturellement, les résultats précédents ne sont plus valables si le passage d'une conformation à l'autre implique une barrière énergétique du même ordre de grandeur que celle des réactions dans lesquelles elles sont impliquées. Notons qu'il existe des cas où la différence d'énergie est telle qu'on peut séparer les conformations. C'est le cas par exemple pour des atropisomères.
Le principe de Curtin-Hammett trouve aussi une illustration dans le dédoublement cinétique dynamique.
Les types d'inversion stéréochimiques
Racémisation
Il s'agit de la transformation qui accompagne le passage d'un composé énantiopur au mélange racémique des deux énantiomères. Elle s'accompagne
d'une diminution du pouvoir rotatoire de la solution, de la valeur caractéristique du composé pur jusqu'à zéro.
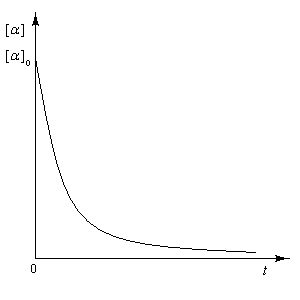
Un exemple de racémisation est fourni par l'énolisation d'une cétone dont l'atome de carbone en a est un centre chiral.
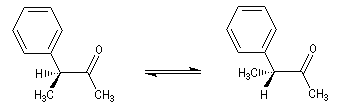
Un autre exemple est constitué par les substitutions nucléophiles monomoléculaires sur les dérivés halogénés ou sur les alcools.
|
|
La molécule ci-dessous représente l'énantiomère (-) de la thalidomide. Le centre chiral possède une configuration absolue S. La molécule a une action sédative non toxique. Cependant ce centre chiral n'est pas stable et in vivo, on observe une racémisation qui conduit à l'énantiomère R tératogène.
|
Epimérisation, anomérisation
Il s'agit de la transformation qui accompagne la mise en équilibre de deux épimères. C'est donc une interconversion entre des diastéréo-isomères particuliers. Puisque les diastéréo-isomères sont chiraux ou non, cette transformation peut ou non s'accompagner d'une modification du pouvoir rotatoire de la solution.
Un exemple est fournit par l'épimérisation en C2 des aldohexoses qui met en jeu la tautomérie céto-énolique.
On rencontre aussi ce type de transformation dans l'hémiacétalisation des sucres. On préfère parler dans ce cas d'anomérisation pour distinguer cette épimérisation particulière dans laquelle la transformation affecte l'atome de carbone de la fonction hémiacétal. Dans ce cas, les molécules sont chirales, et le pouvoir rotatoire passe de la valeur caractéristique des épimères purs (A) ou (B) à celle du mélange en équilibre des deux épimères. L'exemple suivant concerne les hémiacétals cycliques du glucose. Le a-D-glucopyranose et le b-D-glucopyranose.
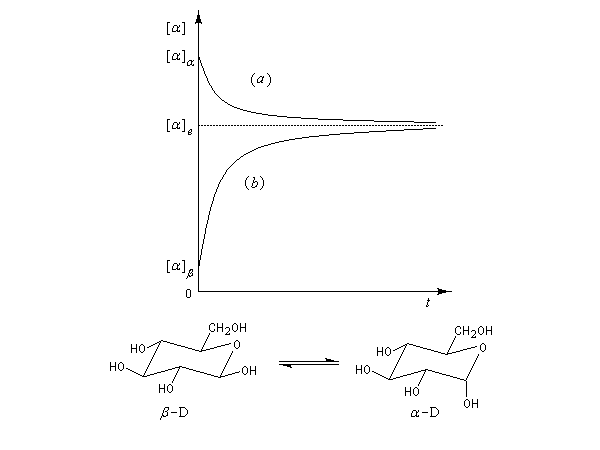
La solution est le siège d'un équilibre qui s'établit par l'intermédiaire de la forme ouverte. L'équilibre d'épimérisation s'accompagne d'un phénomène optique appelé mutarotation. Lorsqu'on dissout dans l'eau l'un des deux composés purs, le pouvoir rotatoire du mélange passe de la valeur caractéristique de ce composé à une valeur correspondant au mélange en équilibre des deux anomères.
|
Composé |
a -D-glucopyranose |
b -D-glucopyranose |
Mélange à l'équilibre |
|
[a] (°.g-1.cm3.dm-1) |
112 |
19 |
52,2 |
Dédoublement cinétique
Le dédoublement cinétique est basé sur le fait que les constituants d'un mélange racémique réagissent avec des vitesses différentes avec un réactif chiral ou au cours d'une réaction énantiosélective catalysée par un catalyseur chiral. Il s'agit donc d'une réaction sous contrôle cinétique. La différence de vitesse des deux réactions provient du fait que les états de transition formés entre le substrat et le catalyseur sont diastéréo-isomères. Le profil énergétique de telles réactions énantiosélectives est étudié dans un paragraphe particulier.
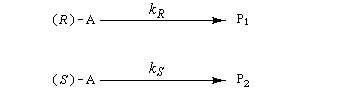
Le facteur de stéréosélectivité est défini comme le quotient des constantes de vitesse :
s = kR/kS (en anglais : relative rate)
Si C0 désigne la concentration totale en A racémique, CR et CS, les concentrations en énantiomères (R)-A et (S)-A respectivement. Le taux de conversion t est défini par la relation :
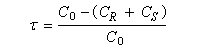
La relation entre ee, t et s a été établie par Kagan dans le cas de réactions de pseudo-premier ordre [18].
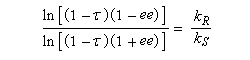
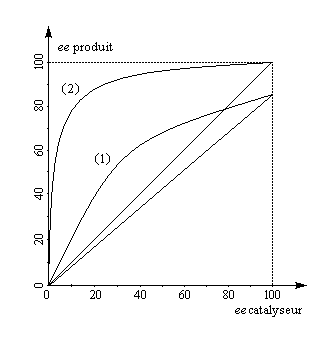
A partir de là, il est possible de tracer les courbes donnant l'excès énantiomérique ee dans le mélange de départ, en fonction du degré de conversion t pour différentes valeur de s. On obtient les graphes suivants [19].
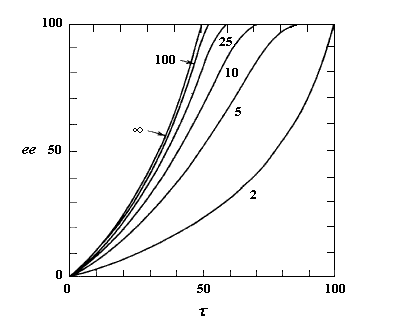
L'excès énantiomérique dans le mélange de départ, qui mesure l'efficacité du dédoublement est d'autant plus grand que :
- le taux de conversion est élevé ;
- le facteur de stéréosélectivité est plus grand.
C'est seulement dans le cas où kR >> kS que l'excès énantiomérique atteint une valeur voisine de 100 % pour un taux de conversion maximal de 50 %.
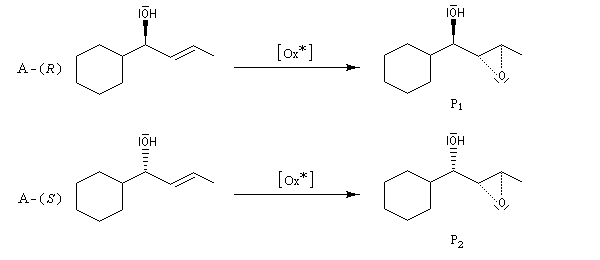
L'époxydation de Sharpless des alcools allyliques a été utilisée dans ce but. La réaction conduisant à P1 est beaucoup plus rapide que celle conduisant à P2 (kR/kS = 140)
Dédoublement cinétique dynamique
Considérons comme précédemment deux énantiomères (R)-A et (S)-A impliqués dans une réaction énantiosélective vis à vis d'un réactif R ou d'un catalyseur chiraux avec des constantes de vitesse différentes kR et kS. Cette fois, les deux énantiomères sont impliqués dans un équilibre de racémisation. Si cet équilibre est suffisamment rapide par rapport à la réaction énantiosélective, le principe de Curtin-Hammett montre qu'on va obtenir majoritairement un seul des deux énantiomères. La séparation peut conduire théoriquement à un rendement de 100 %. C'est le principe de la méthode de dédoublement cinétique dynamique (en anglais : dynamic kinetic resolution DKR).
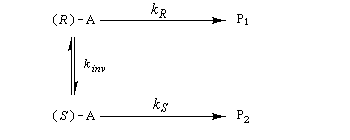
La réaction peut impliquer un réactif chiral pur ou un catalyseur chiral. Dans ce cas il s'agit de catalyse énantiosélective.
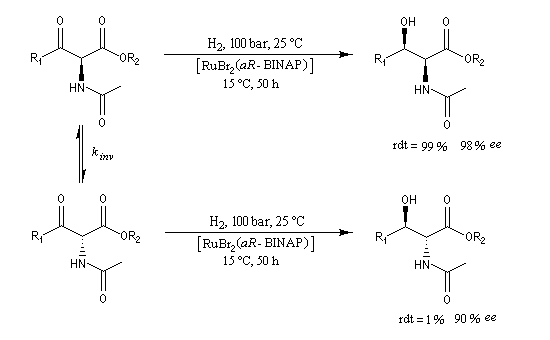
L'hydrogénation énantiosélective des b-céto-esters a été étudié par Noyori [10].
Un exemple récent concerne l'oxydation de Baeyer-Villiger énantiosélective de cétones substituées sous l'action d'un peroxyacide en présence d'un catalyseur chiral. Dans la réaction suivante, la lactone est obtenue avec un rendement de 47 % et un excès énantiomérique de 69 %. Corrélativement, lorsqu'on part d'un mélange racémique, un dédoublement cinétique du mélange s'opère puisque seule la cétone dont la configuration est indiquée est obtenue.
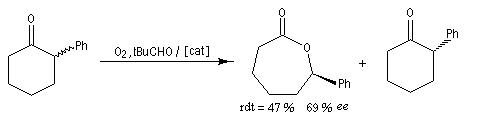
Un autre exemple de dédoublement cinétique utilisant un catalyseur de Jacobsen est donné à la référence [37]
Sélectivité, stéréospécificité
Réactions stéréosélectives
Considérons une réaction au cours de laquelle un substrat A conduit à plusieurs produits stéréoisomères : A1, A2, ... , An. Cette réaction est qualifiée de stéréosélective
si elle conduit de façon préférentielle, voire exclusive, à l'un d'entre-eux :
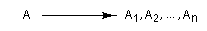
- une réaction diastéréosélective est une réaction au cours de laquelle un ou plusieurs éléments stéréogènes nouveaux sont introduits dans une molécule de manière telle que des diastéréo-isomères sont produits en quantités inégales ;
- une réaction énantiosélective est une réaction au cours de laquelle sont obtenus, à partir d’un précurseur achiral, deux énantiomères d’un produit chiral en quantités inégales.
La maitrise des réactions énantiosélectives constitue un enjeu économique très important dans la synthèse de composés énantiopurs à visée thérapeutique. En 1997, la répartition des molécules de la pharmacopée était la suivante [14] :
- composés énantiopurs 51 % ;
- composés achiraux 32 % ;
- mélanges racémiques 17 %.
Réactions stéréospécifiques
Considérons une réaction de grande stéréosélectivité permettant la transformation d'un substrat A en
un produit A1. La réaction est dite stéréospécifique si un stéréoisomère B de A donne, dans les
mêmes conditions, un produit B1, stéréoisomère de A1. Autrement dit, dans une réaction stéréospécifique, la stéréochimie des produits est déterminée par celle du substrat.
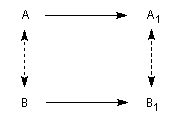
- Une réaction diastéréospécifique fournit des produits diastéréo-isomères lorsqu'elle est réalisée à partir de deux substrats qui ne diffèrent que par leur configuration relative.
Exemple : l'hydrogénation catalytique sur Ni de Raney du (Z)-3,4-diméthylhex-3-ène de configuration Z fournit le (3R, 4S)-diméthylhexane (1') (composé unlike) à l'exclusion du couple d'énantiomères
(3R, 4R)-diméthylhexane et (3S, 4S)-diméthylhexane tous deux diastéréo-isomères du précédent.
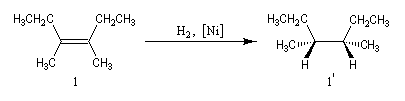
Dans les mêmes conditions, l'hydrogénation du (E)-3,4-diméthylhex-3-ène de configuration E fournit les énantiomères (3R, 4R)-diméthylhexane et (3S, 4S)-diméthylhexane 2' et 2''
(couple like) à l'exclusion du (3R, 4S)-diméthylhexane.
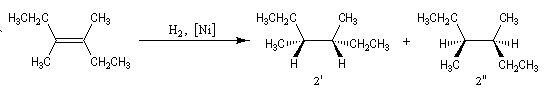
Un autre exemple de réaction diastéréospécifique est fourni par l'élimination bimoléculaire E 2 sur les dérivés halogénés.
- Une réaction énantiospécifique fournit des produits énantiomères à partir de deux substrats qui ne diffèrent que par leur configuration absolue.
Exemple : lorsqu'on effectue la réaction entre le (1S)-1-bromo-1-deutéroéthane de configuration absolue S (1) et NaI dans l'acétone, on obtient
le (1R)-1-iodo-1-deutéroéthane (1') à l'exclusion de son énantiomère le (1S)-1-iodo-1-deutéroéthane.
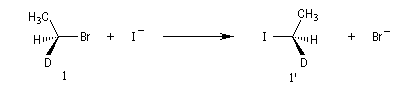
Dans les mêmes conditions, le (1R)-1-bromo-1-deutérobutane (2) fournit le (1S)-1-iodo-1-deutéroéthane (2').
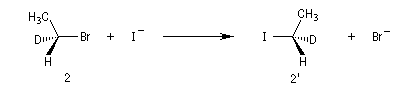
L'inversion de configuration du centre chiral
porte le nom d'inversion de Walden.
Remarque 1 : l'inversion de Walden peut naturellement s'effectuer sans modification de la configuration absolue. Pour un exemple de ce type.
Remarque 2 : la stéréospécificité peut être totale ou partielle. Dans ce cas, il y a formation préférentielle de stéréoisomères.
Remarque 3 : une réaction stéréospécifique est toujours stéréosélective mais l'inverse n'est pas vrai. L'hydrogénation
d'un alcyne catalysée par le palladium de Lindlar est une réaction fortement diastéréosélective de stéréochimie syn.
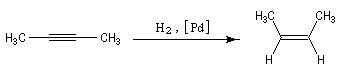
Elle n'est pas stéréospécifique car il n'y a pas de stéréoisomère du substrat.
Rapport diastéréoisomérique, excès diastéréoisomérique
Considérons un mélange de diastéréo-isomères A1 et A2 de concentrations respectives C1 et C2. On adopte les définitions suivantes :
- le rapport diastéréo-isomérique (en anglais : diastereoisomeric ratio) la quantité (C2 < C1) :
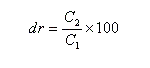
- l'excès diastéréo-isomérique (en anglais : diastereoisomeric excess) est défini par :
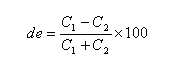
Ces notions s'appliquent en particulier à une réaction diastéréosélective qui transforme un substrat A en deux produits diastéréo-isomères A1 et A2.
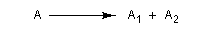
Excès énantiomérique, rapport énantiomérique
Considérons une solution constituée d'un mélange d'énantiomères A1 et A2. Faisons l'hypothèse que l'un des énantiomères, par exemple A1 est prépondérant. Désignons respectivement par C1 et C2 les concentrations des énantiomères A1 et A2.
On appelle rapport énantiomérique (en anglais : enantiomeric ratio), la quantité (C2 < C1) :
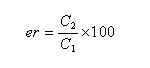
J. Morrisson et S. Mosher, ont introduit l'excès énantiomérique ee (en anglais : enantiomeric excess) :
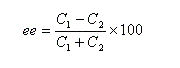
- si l'énantiomère A1 est pur : ee = 100 ;
- si le mélange est un mélange racémique de A1 et de A2 : ee = 0.
Ces grandeurs ne sont pas indépendantes. En effet, en combinant les relations précédentes, on obtient :
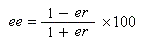
Ainsi que l'a fait remarquer Kagan, l'utilisation du rapport énantiomérique est plus commode que celle de l'excès énantiomérique lorsqu'on cherche à comparer des vitesses relatives de réaction.
L'une méthodes les plus simples qui sont utilisées pour mesurer les excès énantiomériques est la polarimétrie. Dans le paragraphe consacré aux méthodes chiroptiques, on a introduit la notion de pureté pureté optique po grandeur adaptée à la mesure du pouvoir rotatoire. Lorsque la loi d'additivité est applicable (ce qui suppose l'absence de phénomènes non linéaires tels que l'effet Horeau) l'excès énantiomérique (ee) est égal à la pureté pureté optique.
Une autre technique largement employée est l'utilisation d'agents chiraux de dérivatisation. L'idée est de substituer à la mesure de l'excès énantiomérique celle d'un excès diastéréoisomérique.
Le principe est schématisé sur le dessin ci-dessous :
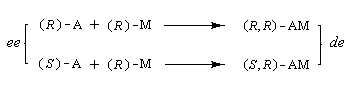
L'agent de dérivatisation est un composé chiral énantiomériquement pur symbolisé ici par M. Par réaction avec les énantiomères à étudier, on forme des diastéréo-isomères dont les propriétés physiques sont différentes.
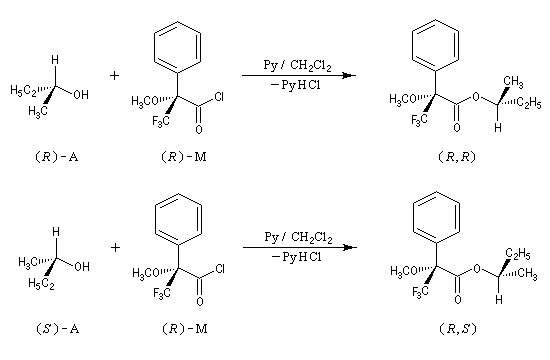
Le plus connu de ces réactifs, l'acide de Mosher ou acide a-méthoxy-a-trifluorophénylacétique (MTPA), fournit des diastéréo-isomères qui sont en général faciles à distinguer en RMN du 1H ou du 19F.
Dans l'exemple ci-dessous, c'est le chlorure d'acyle, du MTPA qui est utilisé car il est plus réactif que le MTPA lui même.
Cela est possible si :
- la transformation est totale ;
- on n'observe pas de dédoublement cinétique qui provoquerait une modification de la composition du mélange.
On aura alors : ee = de
Ces notions s'appliquent en particulier à une réaction énantiosélective qui transforme un substrat A en deux produits énantiomères A1 et A2.
Synthèse stéréosélective de composés chiraux
Synthèse asymétrique, induction asymétrique
La synthèse stéréosélective d'un composé chiral à partir d'un précurseur achiral sans recours à des dédoublements de produits ou d'intermédiaires racémiques est appelée traditionnellement synthèse asymétrique.
Il existe un désaccord sur l'extension de cette définition à des substances dont les molécules comportent déjà un ou plusieurs éléments chiraux, et dans lesquelles la synthèse introduit un nouvel élément chiral. C'est la raison pour laquelle il est préférable de remplacer ce terme traditionnel par synthèse stéréosélective et plus précisément par synthèse énantiosélective ou synthèse diastéréosélective selon le cas.
Le terme d'induction asymétrique se réfère à la formation prédominante de l’un des énantiomères ou diastéréo-isomères possibles au cours d’une réaction sous l’influence d’un facteur chiral.
On distingue, par ordre chronologique, plusieurs générations de méthodes mises au point et employées dans la synthèse de composés chiraux :
- utilisation d'un substrat de départ appartenant au fond chiral (en anglais : chiral pool). Il s'agit donc ici plutôt d'hémisynthèse ;
- un auxilliaire chiral est lié temporairement au substrat de façon covalente puis détaché de celui-ci en fin de réaction ;
- le substrat chiral réagit avec un réactif chiral ;
- le substrat prochiral réagit avec un réactif achiral en présence d'un catalyseur chiral.
Molécules appartenant au fonds chiral
On appelle fonds chiral (en anglais : chiral pool) l'ensemble des molécules chirales d'origine naturelle.
- aminoacides ;
- aminoalcools ;
- hydroxyacides ;
- terpènes ;
- sucres ;
- les alcaloïdes sont des composés azotés hétéroycliques basiques.
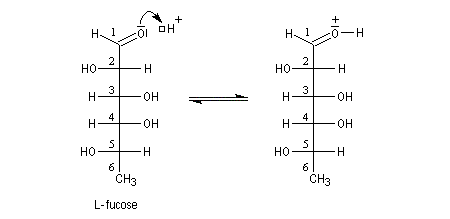
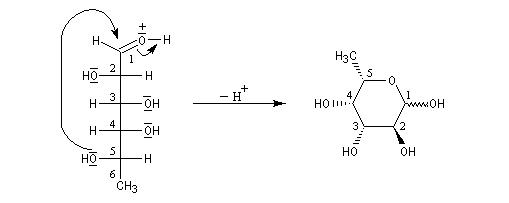
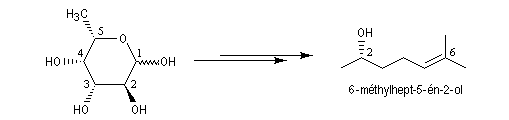
Une synthèse du 6-méthylhept-5-én-2-ol, une phéromone d'insecte encore appelée sulcatol, part du L-fucose comme substrat chiral de départ. La cyclisation du L-fucose en hémiacétal permet la conservation de 4 centres chiraux. Notons qu'un seul d'entre-eux (le C5) est conservé dans la molécule cible.
Aspect énergétique
Il faut distinguer le cas d'une synthèse diastéréosélective de celui d'une synthèse énantiosélective.
- Dans une synthèse diastéréosélective, les produits n'ont pas la même enthalpie libre de référence. On distingue deux cas selon que le produit cinétique (le plus vite formé) est, ou non, différent du produit thermodynamique (le plus stable). Cela est illustré ci-dessous sur l'exemple de la préparation
de deux couples de molécules P1 et P2 possédant deux centres chiraux à partir d'un même substrat A. ;
- dans une synthèse énantiosélective, les produits ont la même enthalpie libre. Pour qu'un excès énantiomérique soit observé il faut opérer sous contrôle cinétique avec des états de transition diastéréo-isomères dont les enthalpies libres sont différentes.
Dans l'exemple ci-dessous, la formation à partir du même substrat A de l'énantiomère P1 (-) est plus rapide que celle de l'énantiomère P2 (+).
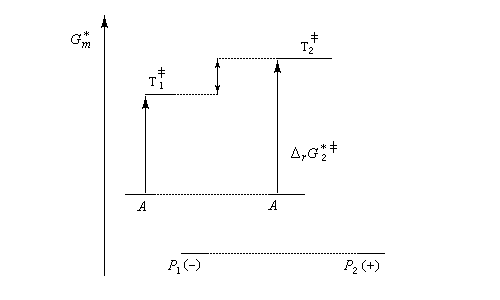
Ce dernier cas se rencontre dans le dédoublement cinétique d'un mélange racémique.
Réactions contrôlées par le substrat
Introduction
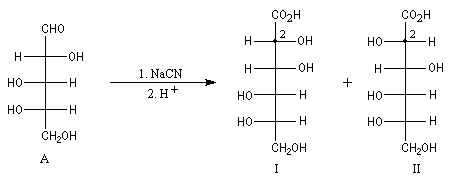
Un cas particulier d'induction asymétrique est l'influence exercée par un centre chiral sur le déroulement stéréochimique d'une réaction impliquant les faces diastéréotopiques d'une fonction insaturée située à proximité de ce centre chiral. Un exemple typique est l'influence exercée par un atome de carbone asymétrique adjacent à un groupe carbonyle. Les premiers exemples de ce type remontent aux travaux d'Emil Fischer lors de son étude des sucres de la famille du glucose (1894). Par addition d'ions cyanure suivie d'hydrolyse sur le L-arabinose, Fischer avait remarqué que seul l'acide mannonique I est obtenu à l'exclusion de son épimère, l'acide gluconique II.
Il s'agit dans ce cas d'induction asymétrique 1,2 car le centre asymétrique inducteur est lié à celui qui est nouvellement créé.
Notons que le contrôle de la diastéréosélectivité en série acyclique est souvent plus difficile à atteindre qu'en série cyclique. En effet, il existe une beaucoup plus grande liberté conformationnelle dans le premier cas que dans le second. Cependant, des progrès spectaculaires ont été réalisés ces dernières années et le contrôle stéréochimique en série acyclique est de plus en plus souvent mis à profit en synthèse.
Règle de Cram
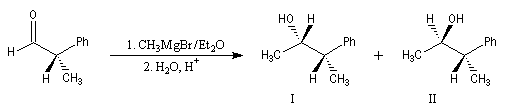
Raisonnons sur l'exemple de l'addition d'un réactif organométallique sur un substrat carbonylé. La molécule de départ est la (2R)-2-phénylpropanal.
La molécule comporte un atome de carbone asymétrique adjacent au groupe carbonyle. La présence de ce centre chiral
rend les faces du carbonyle diastéréotopiques. L'addition d'un nucléophile achiral
sur chacune de ces faces conduit à deux composés diastéréoisomères 1 et 2 en proportions différentes. Il s'agit d'une réaction diastéréosélective.
|
Composé |
I |
II |
|
% de produit |
71 |
29 |
En 1952 le chimiste américain D. J. Cram a proposé un modèle empirique d'état de transition permettant de prévoir dans un certain nombre de cas lequel des deux diastéréoisomères est le plus abondant. Cela revient à rechercher laquelle des deux faces prochirales réagit préférentiellement
avec le composé organométallique. Les groupes liés à l'atome de carbone asymétrique sont classés selon leur taille :
G > M > P
Le groupe G est placé le plus loin possible du groupe carbonyle c'est à dire dans une conformation antipériplanaire. La règle de Cram s'appuie sur une base stérique. Le nucléophile va réagir de façon préférentielle sur la face du carbonyle la plus dégagée c'est
à dire celle qui contient le groupe le moins volumineux (P).
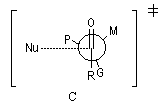
Cette règle empirique permet de rendre compte des résultats obtenus plus haut.
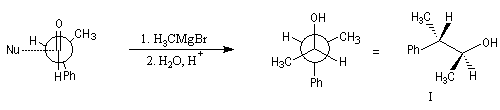
Plusieurs modèles d'états de transition ont été proposés pour rendre compte de l'induction asymétrique 1, 2. On va examiner successivement le modèle de Felkin puis une amélioration de ce dernier appelé modèle de Felkin-Anh.
Modèle de Felkin
H. Felkin (1968) a proposé une justification de la règle de Cram à partir de considérations d'analyse conformationnelle.
Les groupes liés à l'atome de carbone asymétrique sont classés par taille décroissante :
G > M > P
Le carbonyle est placé perpendiculairement à G, tandis que le groupe R est placé le plus loin de M. Le nucléophile attaque le carbonyle dans une direction perpendiculaire. Cela correspond aux situations suivantes :
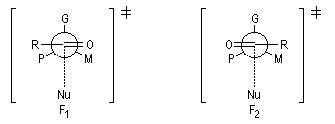
A ces états de transition correspondent deux produits diastéréoisomères. L'état de transition 1 est favorisé à cause de l'interaction moindre entre P et R qu'entre M et R.
Ce modèle pose un problème dans le cas des aldéhydes.
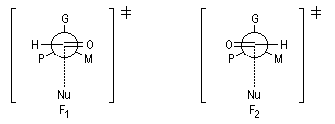
L'état de transition le plus favorable prévu est F2. Mais l'expérience montre que le produit attendu n'est pas majoritaire.
Modèle de Felkin-Anh
Le modèle de Felkin-Anh (1976), propose une approche plus fine que dans le modèle de Felkin. Dans ce dernier, le choix entre les états de transition F1 et F2 repose seulement sur les interactions au sein du substrat car toutes les distances faisant intervenir le nucléophile sont identiques. Dans le cas du modèle de Felkin-Anh le nucléophile intervient. Il réagit avec le carbonyle dans le demi-espace contenant P en respectant l'angle de Dunitz-Bürgi voisin de 105° (ou inférieur à cette valeur selon qu'il y a ou non assistance électrophile.) L'état de transition ci-dessous est favorisé car la trajectoire du nucléophile est celle qui passe au plus près du groupe P.
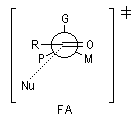
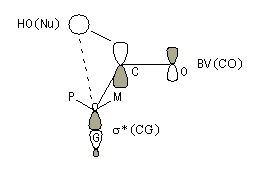
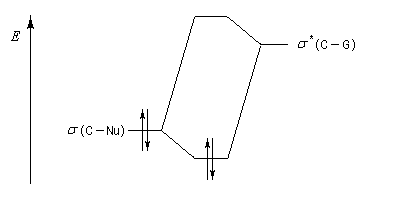
Mais ce modèle ne se base pas uniquement sur des considérations stériques. Il fait intervenir le recouvrement des orbitales du substrat et celles du nucléophile. En termes d'orbitales frontières, la réaction implique le recouvrement entre la plus haute orbitale occupée du nucléophile et la plus basse orbitale vacante du carbonyle.
|
|
Justification orbitalaire du modèle de Felkin-Anh. Lors de l'approche du nucléophile, les deux faces du carbonyle ne sont pas équivalentes. En plus de l'interaction principale entre la plus haute orbitale occupée par le nucléophile (HO) et la plus basse orbitale vacante (BV) du carbonyle (trait plein), la disposition perpendiculaire entre les liaisons CO et CG permet un recouvrement secondaire favorable (pointillés) entre la HO du nucléophile et l'orbitale antiliante s* de la liaison C-G (G est le groupe le plus "volumineux" ; voir plus bas.) [7]. Il s'agit donc d'un effet d'hyperconjugaison.
|
Le classement des substituants dans la suite : G > M > P n'est pas toujours facile à effectuer. Deux critères interviennent :
- l'encombrement effectif peut souvent être estimé au moyen des rayons de Van der Waals (mais pas toujours) ;
- le caractère attracteur du groupe lié à son caractère électronégatif.
A titre d'exemple un atome comme Cl "l'emporte" sur un groupe tBu.
Dans l'exemple suivant, NBn2 qui désigne le groupe amino protégé par un groupe benzyle, est traité comme un groupe G.
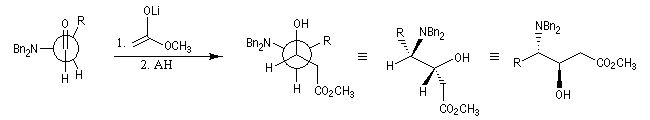
Il existe d'autres explications de ce phénomène. Un modèle électrostatique élémentaire (Wipf) consiste à admettre que le nucléophile va réagir du côté positif du dipôle formé par la liaison carbone-hétéroatome électronégatif. Donc, comme il le ferait avec un groupe volumineux.
Pour une mise au point détaillée, à un niveau avancé, des différents modèles : voir [49]
Angle de Flippin-Lodge
Considérons la réaction suivante :
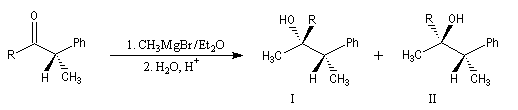
La diastéréosélectivité augmente avec l'encombrement du groupe R
|
R |
I (Felkin) |
II (anti-Felkin) |
|
H |
71 |
29 |
|
Et |
86 |
14 |
|
tBu |
96 |
4 |
Effet Cieplak
Opinion de D. A. Evans à propos de l'effet Cieplak :
"Structures are stabilized by stabilizing their highest energy filled states. This is one of the fundamental assumptions in frontier molecular orbital theory. The Cieplak hypothesis is nonsense." Prof. David A. Evans
Mise au point sur l'effet Cieplak : [48].
Contrôle par chélation
La réaction suivante fournit avec une grande stéréosélectivité le stéréoisomère II (composé anti-Felkin).
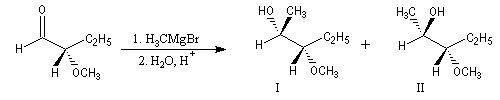
|
Composé |
I (Felkin) |
II (anti-Felkin) |
|
% de produit |
5 |
95 |
Ces résultats s'interprètent par la formation d'un complexe entre l'atome d'oxygène du groupe méthoxy jouant le rôle de base de Lewis et le composé organométallique jouant le rôle d'acide de Lewis.
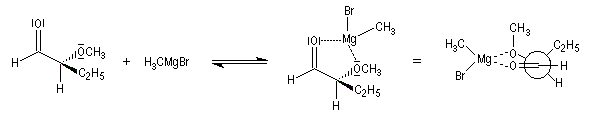
L'attaque du nucléophile s'effectue sur la face la plus dégagée de ce complexe.
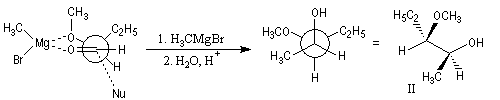
L'application sans discernement du modèle de Felkin, prédirait la formation du diastéréoisomère I du composé précédent.
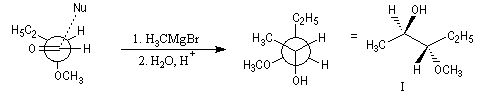
Les cations permettant la chélation sont : Li+, Mg2+, Cu2+, Zn2+, Ti4+. En revanche Na+ et K+,sont en général impropres à jouer ce rôle.
Le contrôle par chélation n'est pas possible lorsque la fonction éther comporte des groupes volumineux tels que TBS ou TBDPS dans les éthers de silyle.
La réaction suivante permet la préparation de diols 1,2 (après déprotection) avec une stéréochimie anti lorsque R est le groupe benzyle (Bn) et une stéréochimie syn lorsque c'est le groupe tertiobutylsilyle (TBS).
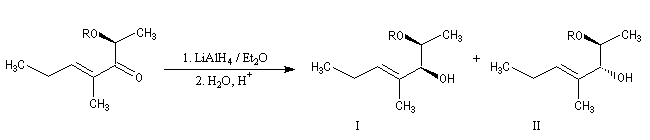
Dans le tableau ci-dessous, on compare les groupes benzyle (Bn) et tertiobutylsilyle (TBS).
|
Composé |
I (Felkin) |
II (chélation) |
|
R = Bn |
2 |
98 |
|
R = TBS |
95 |
5 |
Le phénomène de chélation peut s'accompagner d'une excellente diastéréosélectivité, souvent bien meilleure que celle obtenue dans le cadre d'un contrôle de la stéréochimie selon Felkin-Anh.
remarque : les réactions d'addition nucléophile des hydrures sur le groupe carbonyle font l'objet d'un paragraphe spécifique dans le chapitre relatif aux composés carbonylés.
Règle de Prelog
La règle de Prelog est utilisée pour prévoir le sens de l'addition sur les faces prochirales d'un groupe carbonyle située en g d'un centre chiral [12]. L'exemple suivant concerne l'addition d'un organomagnésien sur un cétoester. Les groupes carbonyles sont placés en position antiparallèle. Le groupe le plus volumineux (G) est placé dans le plan de figure.
L'organométallique attaque le carbonyle dans le demi-plan contenant le groupe le moins volumineux (P).
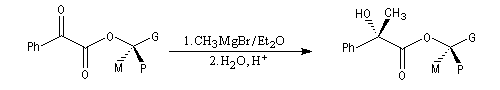
La règle de Prelog a été mise à profit pour déterminer la configuration absolue d'alcools chiraux comme le menthol [12]. Celle-ci est déduite de la configuration de l'acide 2-hydroxy-2-phénylpropanoïque (atrolactique) obtenu après hydrolyse.
Synthèses diastéréosélectives en série cyclique
L'exemple choisi concerne l'addition des hydrures non encombrés sur les cétones cycliques. Elle s'effectue de façon préférentielle
par le biais d'une attaque axiale de l'ion hydrure. Après hydrolyse, l'alcool majoritaire est celui dont le groupe -OH et situé en position équatoriale.
La 4-tertiobutylcyclohexanone fournit un bon modèle pour cette réaction car le
basculement conformationnel est bloqué du fait de la présence du volumineux groupe
tertiobutyle. Les résultats sont les suivants :
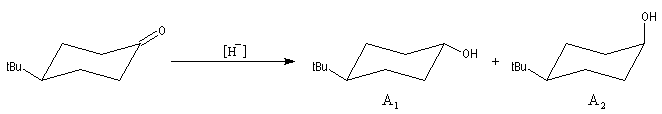
|
Composé |
A1 |
A2 |
|
LiAlH4 |
92 |
8 |
|
LiBH(secBu)3 |
7 |
93 |
|
|
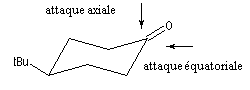
- avec un petit nucléophile, l'attaque préférentielle est axiale ;
- avec un gros nucléophile, l'attaque est de préférence équatoriale de façon à minimiser les interactions 1,3-diaxiales.
|
Le rapport diastéréoisomérique de la réaction est : dr = 90 : 10 tandis que l'excès diastéréoisomérique est : de = 80 %.
Synthèses diastéréosélectives mettant à profit un auxilliaire chiral
Généralités
La synthèse stéréosélective à partir de substrats et de réactifs achiraux est possible à condition d'utiliser
un auxilliaire chiral. Ce dernier est lié de façon covalente au substrat puis détaché de ce dernier. Les avantages sont :
- la formation de diastéréoisomères facilement séparables ;
- une diastéréosélectivité généralement élevée.
Les inconvénients inhérents à ce genre de synthèses sont :
- la fixation de l'auxilliaire sur le substrat puis son enlèvement ajoutent deux réactions supplémentaires à la synthèse ;
- la stéréosélectivité n'est pas toujours facile à prévoir.
Alkylation diastéréosélective d'Evans
La préparation de dérivés acylés d'oxazolidinones substituées a été vue dans le chapitre consacré aux dérivés d'acides. Très récemment, des oxazolidonones chirales ont été préparées avec de très bons excès énantiomériques en mettant à profit la réaction d'aminohydroxylation énantiosélective de Sharpless. Par action du LDA, on obtient essentiellement l'énolate Z.

L'alkylation de l'énolate s'effectue sur la face la plus dégagée, c'est à dire du côté opposé au substituant fixé en 4.
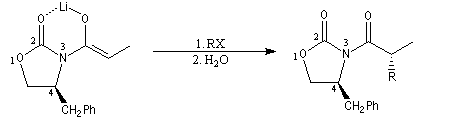
Pour achever la synthèse, il faut détacher l'auxilliaire chiral. La réaction de réduction par LiAlH4 fournit un alcool chiral.
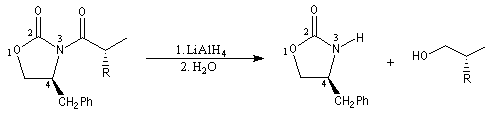
|

|
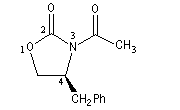
La molécule représentée à gauche est dérivée d'une oxazolidinone d'Evans. Sur le modèle en 3D, on constate que les deux faces ne
possèdent pas le même encombrement.
|
Alkylation diastéréosélective par la méthode RAMP-SAMP
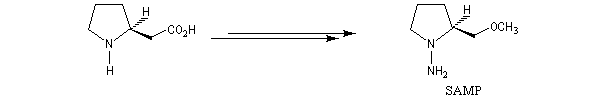
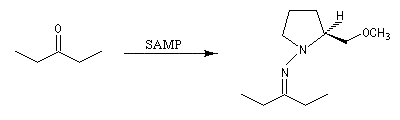
Cette méthode due à D. Enders est basée sur la formation d'hydrazones à partir de composés carbonylés [33].
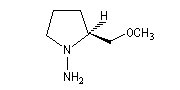
SAMP est le sigle désignant la (S)-1-amino-2-méthoxyméthylpyrrolidine. Son énantiomère, la (R)-1-amino-2-méthoxyméthylpyrrolidine est noté RAMP. Le SAMP est préparé à partir de la (S)-hydroxyméthylpyrrolidine et le RAMP à partir de l'acide glutamique naturel [35].
|
|
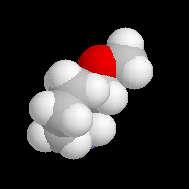
Molécule de (S)-1-amino-2-méthoxypyrrolidine (SAMP) .Sur le modèle en 3D, on constate que les deux faces du cycle ne
possèdent pas le même encombrement.
|
La méthode permet l'alkylation en a des composés carbonylés : aldéhydes et cétones.
L'exemple suivant concerne la synthèse d'une substance de défense d'une araignée ("Daddy-long-legs Spider"). L'excès énantiomérique est supérieur à 95 %.
- formation de l'hydrazone ;
- métallation par le LDA à basse température ;
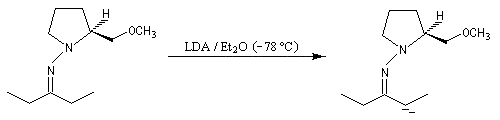
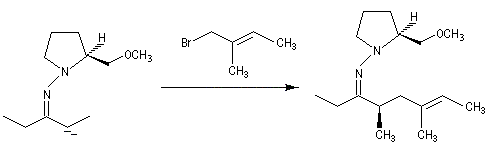
- alkylation de l'anion ;
- clivage de l'intermédiaire.
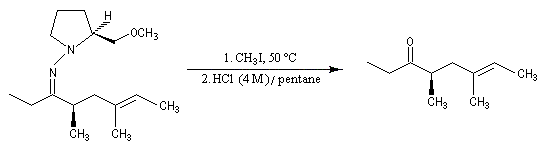
Les avantages de la méthode sont les suivants :
- RAMP et SAMP sont des produits commerciaux, donc facilement accessibles ;
- la réaction est généralisable à pratiquement tous les carbonylés sauf ceux qui présentent un encombrement très important ;
- la déprotection est facile.
Le principal inconvénient est le risque d'épimérisation lors de l'étape de décrochage. Plusieurs méthodes ont été mises au point :
- iodure de méthyle dans HCl ce qui exclut la présence d'un groupe amino ;
- utilisation de l'ozone mais cela exclut la présence de liaisons éthyléniques qui sont clivées par l'ozone.
Transfert temporaire de chiralité
Dans cette méthode due à D. Seebach [15], on cherche à réaliser la transformation suivante.
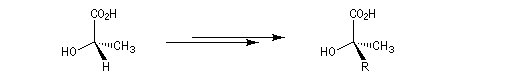
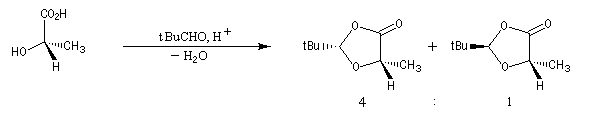
La première étape est la réaction entre la fonction alcool et un aldéhyde encombré. Elle fournit un acétal. Compte tenu de la proximité de la fonction acide carboxylique, la cyclisation fournit une lactone avec une diastéréosélectivité 4 : 1.
Le proton en a du groupe carbonyle du diastéréo-isomère majoritaire est arraché par le LDA pour conduire à un énolate.
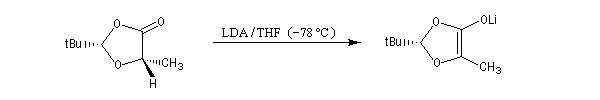
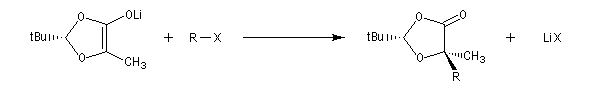
On effectue ensuite l'alkylation de l'énolate par un dérivé halogéné. Le groupe R se fixe sur la face la plus dégagée c'est à dire celle opposée au volumineux groupe tBu.
L'hydrolyse de l'ester permet d'obtenir le produit cherché.
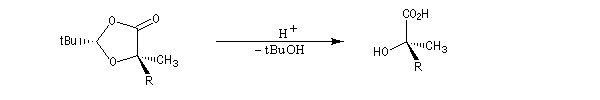
Synthèses énantiosélectives utilisant un réactif chiral
Généralités
La chiralité est introduite via le réactif. Il n'y a plus besoin des étapes de fixation et de décrochage de l'auxilliaire.
Hydrures modifiés
L'alpine borane (réactif de Midland symbolisé par AB) ou le chloroborane de Brown sont des réactifs appartenant à la famille des hydrures de bore neutres (tout comme BH3 ou le DIBAL pour les hydrures d'aluminium). Ils permettent des réductions énantiosélectives de composés carbonylés.
L'exemple suivant concerne la réduction du groupe carbonyle d'un cétoester. L'atome d'hydrogène est transféré sur la face prochirale re du groupe carbonyle conduisant au composé de configuration absolue S avec un excès énantiomérique de 100 %.
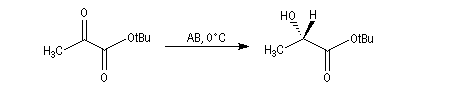
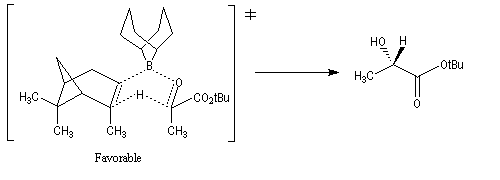
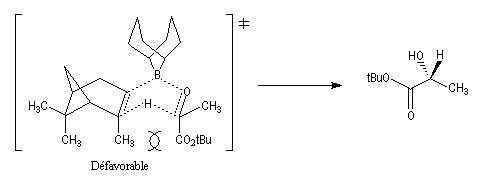
La stéréosélectivité tire son origine du fait que l'hydrogène en b du bore est transféré sur le composé carbonylé de manière à minimiser les interactions 1,3-diaxiales qui apparaissent dans l'état de transition ainsi que l'indique le schéma ci-dessous.
Un autre exemple appartenant à la même catégorie concerne l'addition du diisocamphéylchloroborane (+)-(IPC)2BCl (chloroborane de Brown) sur
le groupe carbonyle prochiral d'une cétone.
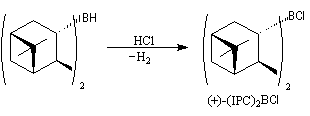
L'addition du (+)-(IPC)2BCl s'effectue de façon préférentielle sur la face prochirale si de la cétone, conduisant à l'alcool de configuration absolue R.
L'origine de l'énantiosélectivité peut être interprétée par le modèle suivant de l'état de transition.
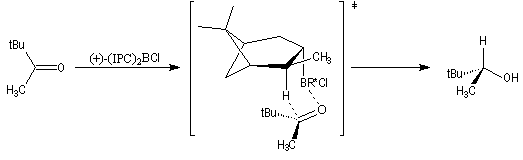
L'addition s'effectue de façon à ce que le groupe le plus volumineux lié au carbonyle, ici le groupe tertiobutyle, soit le plus éloigné du groupe méthyle du réactif.
Hydroboration-oxydation
Ce sujet a été étudié dans le chapitre consacré aux composés éthyléniques.
Le diisopinocamphéylborane Ipc2BH est le premier réactif de ce type introduit par H. C. Brown en 1961. Il permet de bons excès énantiomériques à partir de composés éthyléniques de stéréochimie cis.
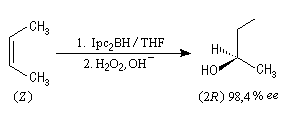
Il existe d'autres agents d'hydroboration, comme le (trans)-diméthyl-2, 5-diméthylborolane proposé par S. Masamune en 1985.
Déprotonation énantiosélective
L'utilisation d'une base chirale dérivée de la (S)-proline a permis au chimiste japonais M. Asami d'obtenir de très grands excès énantiomériques pour la déprotonation d'époxydes méso [16]. Dans l'époxyde suivant, qu'on peut considérer comme un dérivé du cyclohexène, les atomes d'hydrogène Ha et Hb sont énantiotopiques.
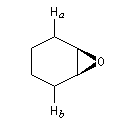
Par ouverture énantiosélective, l'alcool allylique de configuration absolue S est obtenu avec un rendement de 77 % et un excès énantiomérique de 79 %.
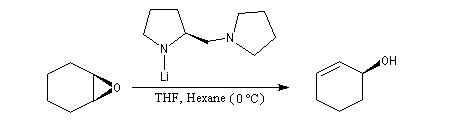
Ce résultat peut être rationalisé en considérant les états de transition suivants.
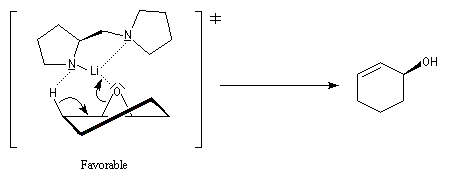
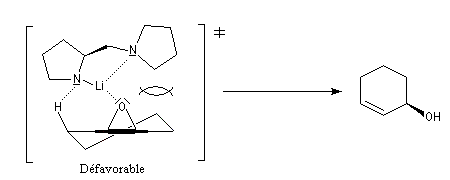
Ce type de réaction a été utilisé avec succès dans la synthèse du Lasiol (Kasai, 1993).
Un autre exemple de déprotonation énantiosélective concerne la réaction entre le butyllithium complexé par la spartéine sur un dérivé de la pyrrolidine.
Catalyse énantiosélective
Généralités
Le catalyseur intervient dans l'état de transition afin de disposer réactif et substrat dans une géométrie préférentielle. Cette façon de procéder offre beaucoup d'avantages par rapport aux méthodes précédentes. Seul le catalyseur est nécessairement chiral. Il est utilisé en faible quantité et il peut être facilement séparé du milieu réactionnel par chromatographie. Le premier exemple de ce type fut rapporté par Betti et Luchi en 1940. Il s'agissait de la réaction entre un composé organométallique, l'iodure de méthylmagnésium et le benzaldéhyde en présence de N, N-diméthylbornylamine.
La première réaction stéréosélective utilisant un catalyseur métallique chiral soluble permettant de discriminer les faces prochirales d'un substrat a été rapportée par R. Noyori en 1966 [13].
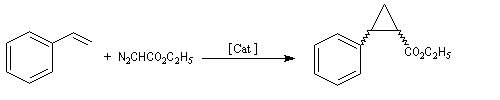
La formule du catalyseur utilisé est donnée ci-dessous.
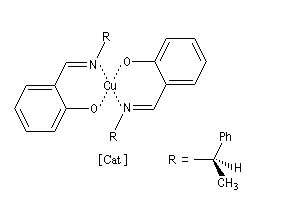
Pour une revue détaillée sur le sujet, voir par exemple : [52].
Hydrogénation énantiosélective
Voici l'exemple de la synthèse d'un précurseur des vitamines E et K à partir du nérol, un alcool allylique. L'excès énantiomérique est : ee = 98 % [34] et [36-c].
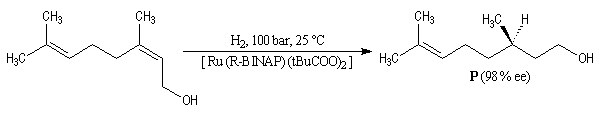
|
|
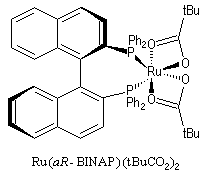
Le ligand BINAP introduit par R. Noyori, existe sous deux formes énantiomères atropisomères. Le aR-BINAP est l'un des constituants
d'un catalyseur homogène au ruthénium utilisé dans les réactions d'hydrogénation catalytique énantiosélectives.
|
L'hydrogénation énantiosélective est utilisée comme réaction permettant le dédoublement cinétique dynamique de mélange racémiques d'énantiomères.
Un autre exemple d'utilisation de l'hydrogénation énantiosélective catalysée par un complexe chiral soluble est celui de la synthèse par W. S. Knowles de la (L)-DOPA,
un médicament actif vis à vis de la maladie de Parkinson (voir par exemple : [36-c].)
Isomérisation énantiosélective
Les catalyseurs de Noyori peuvent également être utilisés pour promouvoir des réactions d'isomérisation. Un exemple est fourni par une étape de la synthèse industrielle du (-)-menthol à partir du myrcène, selon un procédé mis au point par des chercheurs de la firme japonaise Takasago [32]. Ce procédé fournit actuellement environ 30 % du marché mondial du (-)-menthol.
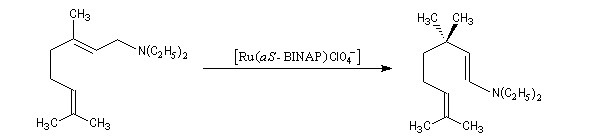
Epoxydation énantiosélective (AE)
- L'époxydation de Sharpless concerne les alcools allyliques.
- L'époxydation de Jacobsen est réalisable sur les composés éthyléniques ordinaires.
Dihydroxylation énantiosélective (AD)
K. B Sharpless 1987. La dihydroxylation énantiosélective des doubles liaisons éthyléniques.
Aminohydroxylation énantiosélective (AA)
K. B Sharpless 1996. L' aminohydroxylation énantiosélective des doubles liaisons éthyléniques.
Réaction de Corey-Bakshi-Shibata
Cette réduction énantiosélective de composés carbonylés prochiraux utilise comme catalyseur une oxazaborolidine comme catalyseur. Elle est étudiée dans le chapitre consacré aux composés carbonylés.
Amplification chirale
La plupart du temps, lors d'une catalyse ou d'une autocatalyse énantiosélectives, l'excès énantiomérique du produit ee (p) est proportionnel à celui du catalyseur ee (c).
ee (p) = eeo.ee (c)
eeo est l'excès énantiomérique du produit lorsque celui du catalyseur vaut 100. Lorsque cette proportionnalité n'a pas lieu, il s'agit d'un effet non linéaire :
- un effet non linéaire positif produit davantage de produit que lorsque la relation de proportionalité s'applique ;
- l'effet non linéaire est négatif dans le cas contraire.
Lorsque l'effet non linéaire positif est élevé, on parle d'amplification chirale. Il s'agit naturellement de l'effet non linéaire le plus intéressant puisqu'il permet d'utiliser un auxilliaire chiral possédant une faible pureté énantiomérique et d'obtenir des rendements énantiomériques élevés en produits.
Le premier exemple d'amplification chirale significative a été mis en évidence par le chimiste français H. B. Kagan à propos de l'époxydation asymétrique de Sharpless du E-géraniol.
Un modèle, proposé par Kagan en 1994 fait intervenir la formation de diastéréoisomères TiLRLS , TiLRLR et TiLSLS catalytiquement actifs avec une constante de vitesse différente, présents dans des proportions fixées par une constante d'équilibre K.
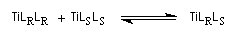
Selon la réactivité du complexe méso TiLRLS par rapport aux complexes chiraux TiLSLS et TiLRLR un effet non linéaire positif ou négatif peut-être observé.
|
|
Un exemple d'amplification chirale mis en évidence par R. Noyori concerne l'addition énantiosélective des dialkylzinciques sur les aldéhydes aromatiques.
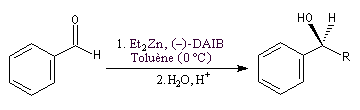
|
Autocatalyse énantiosélective
Dans ce cas, le produit chiral formé sert de catalyseur pour sa propre synthèse. Il est mis en quantité catalytique au début de la synthèse.
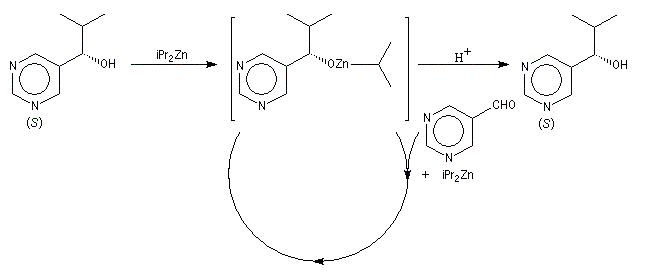
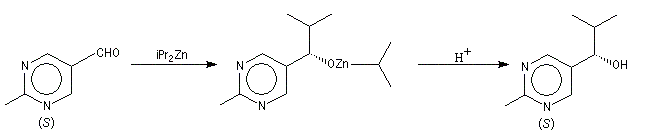
3 mg de substrat présentant un excès énantiomérique ee = 0, 18 % conduisent à 323 mg de produit avec un excès énantiomérique ee = 83, 2 %.
On voit que l'amplification chirale est très importante.
Articles
[1] E. L. Eliel et F. J. Biros, J. Am. Chem. Soc. 88, 3334 (1966).
[2] V. Pellegrin - Les représentations graphiques bidimensionnelles des molécules en chimie organique (Bulletin de l'Union des Physiciens, février 1999).
[3] D. Y. Curtin, Rec. Chem. Prog. 15, 111 (1954).
[4] Unambiguous specification of stereoisomerism about a double bond, J. E. Blackwood et coll. J. am. Chem. Soc., ,90, 509, (1968). (utilisation des descripteurs Z et E)
[5] D. J. Cram et F. A. Abd Elhafez, J. am. Chem. Soc., 74, 5828 (1952).
[6] M. Cherest, H. Felkin, N. Prudent, Tetrahedron lett.,1968, 18, 2199, 2204.
[7] N. T. Anh, O. Eisenstein Tetrahedron lett.,1976, 155 (article dans lequel les auteurs proposent une modification de la règle de Felkin ; ce qui deviendra le modèle de Felkin-Anh.)
(voir aussi le lien : [31] sur le site de D. Evans.)
[8] K. Soai. Nature 1295, 378, 767.
[9] K. Soai. Tetrahedron : Asymmetry, 1997, 8, 1717.
[10] R. Noyori et al J. am. Chem. Soc., 111, 9134-9135 (1989).
[11] R. Noyori et al J. Am. Chem. Soc, 108, 6071, 1986.
[12] Prelog. V, Helv. Chim. Acta. 1953, 36, 308.
[13] Nozaki, H. ; Moriuti, S. ; Takaya, H. ; Noyori, R. , Tetrahedron lett.,1966, 6239.
[14] Chemistry and Engeneering news, octobre 1997.
[15] D. Seebach Tetrahedron 1984, 40, 1313.
[16] M. Asami, T. Ishizaki, S. Inoue, Tetrahedron, Asymmetry, 1994, 5, 5, 793.
[18] H. B. Kagan, J. C. Fiaud. Top. Stereochem. 1988, 18,249-330
[19] Victor S. Martin, S. Woodard, Tsutomu Katsuki, Yasuhiro Yamada, Masonari Ikeda, K. Barry Sharpless, J. Am. Chem. Soc., 1981, 103 (20), pp 6237-6240
Liens
[30] Catalytic Enantioselective Additions of Dialkyl Zincs to Aldehydes using (2S)-DAIB) by Masato Kitamura, Hiromasa Oka, Seiji Suga, and Ryoji Noyori.
[31] Theoritical Interpretation of 1,2 asymmetric induction. The importance of antiplanarity Nguyên Trong Anh ans O. Eisenstein, Laboratoire de chimie théorique, Université de Paris Sud.
[32] (-)-menthol from myrcène Procédé Takasago de synthèse du menthol
[33] Asymmetric syntheses using the SAMP/RAMP method by Dieter Enders, Helmut Kipphardt, and Peter Fey
[34] Asymmetric Hydrogenation of Allylic Alcohols using BINAP-Ru Complex by R. Noyori and Coll
[35] SAMP and RAMP versatile chiral auxiliaries by Dieter Enders, Peter Fey, and Helmut Kipphardt
[36-a] Catalyse asymétrique Tutoriel sur la catalyse asymétrique (I), Imperial College (contient notamment une description de l'époxydation de Sharpless.)
[36-b] Catalyse asymétrique Tutoriel sur la catalyse asymétrique (II), Imperial College
[36-c] Catalyse asymétrique Tutoriel sur la catalyse asymétrique (III), Imperial College (contient notamment une description de l'hydrogénation énantiosélective avec application à la synthèse de la L-DOPA.)
[37] Preparation of s-methylglycidate via hydrolytic kinetic resolution by Christian P. Stevenson, Lars P. C. Nielsen, and Eric N. Jacobsen
[38] Perspectives offertes sur la synthèse asymétrique par H. Kagan
[39] Substitutions nucléophiles énantiosélectives sur des aziridines meso par P. Nury
[40] (R-(-)-10-methyl-1(9)-octal-2-one by G. Revial and M. Pfau
[41] Diastéréosélective aldol condensation by James R. Gage and David A. Evans
[42] Transformation of pseudoephedrine amides into highly enantiomerically enriched aldehydes, alcohols and ketones by Andrew G. Myers, Bryant H. Yang, and Hou Chen.
[43] Asymmetric Synthesis of a-amino acids by the alkylation of pseudoephedrine glycinamide : L-allylglycine and N-boc-L-allylglycine by Andrew G. Myers and James L. Gleason.
[44] Felkin-Anh model and chelation control
[45] Synthese de liaisons CC
[46] Termes généraux de la chimie (Journal Officiel 3 juillet 1996)
[47] Dynamic Kinetic Resolution, Pratical Applications in Synthesis By Valerie Keller, November 1, 2001
[48] The Cieplak effect by Simon Meek
[49] The evolution of Stereochemical Models for CO additions by D. A. Evans, Harvard University.
[50] Camphor as a natural source of chirality in assymetric syntheses by Wolfgang Oppolzer, Université de Genève.
[51] Z(O,R)-enolates and E(O,R)-enolates Pr. Ari Koskinen, Helsinky, University of Technology.
[52] The Advent and Development of the Field of Enantioselective Organocatalysis by D. Mac Millan, University of Princeton.
[53] Thèse S. Malfait
[53] Thèse Delacroix
Ouvrages théoriques
J. March, Advanced organic chemistry, Wiley Interscience.
F. A. Carey, R.J. Sundberg, Advanced organic chemistry, 3d edition (Plenum Press, 1990).
H. Kagan, La stéréochimie organique (PUF, 1975).
J. L Pierre Principes de stéréochimie organique statique (A. Colin, 1971).
André Collet, Jeanne Crassous, Jean-Pierre Dutasta, Laure Guy, Molécules chirales : stéréochimie et propriétés (Editions du CNRS, 2006)
Nguyên Trong Anh, Orbitales frontières : manuel pratique, EDP Sciences, CNRS.
Erick M. Carreira, Lisbet Kvaerno, Classics in Stereoselctive Synthesis, J. Wiley, 2009.
J. Seyden-Penne, Synthèse et catalyse asymétriques, (CNRS éditions, 1994).
R.E. Gawley, J. Aubé, Principles of Asymmetric synthesis, Pergamon, 1996.
K. Mislow, Introduction to stereochemistry (W. A Benjamin, New York).
J. Jacques, La molécule et son double (Hachette, 1992).
Morrison, James D.; Mosher, S. Harry : Asymmetric organic réactions, Prentice-Hall, Englewood Cliff, New Jersey, 1971.
RETOUR AU MENU DU COURS
Vous pouvez, si vous le souhaitez, utiliser le contenu de cette page dans un but pédagogique et non commercial.
Gérard Dupuis - Lycée Faidherbe - LILLE
décembre 2012