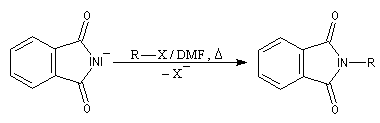Cours de chimie Organique - G. Dupuis - Lycée Faidherbe de Lille
Amides et composés apparentés
Nomenclature
Définitions
Les amides sont divisés en trois classes selon le nombre de groupes acyles portés par l'atome d'azote.
|
Formule |
RCONH2 |
(RCO)2NH |
(RCO)3N |
|
Classe |
primaire |
secondaire |
tertiaire |
Il faut faire attention de ne pas confondre l'appartenance à une classe et le degré de substitution de l'atome d'azote par un groupe alkyle [20]. Les amides primaires peuvent être :
- non substitués à l'azote ;
- monosubstitués (amides N-substitués) ;
- disubstitués (amides N, N-disubstitués.
Amides
- Série acyclique
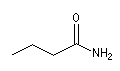
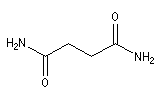
Le nom des amides primaires non susbstitués est obtenu en remplaçant la terminaison oïque de l'acide par la terminaison amide.
|
|
|
|
Butanamide |
Butanediamide |
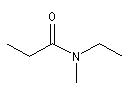
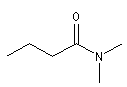
Les amides primaires substitués sur l'atome d'azote sont nommés en faisant précéder le nom de l'amide de la lettre N suivie du nom du groupe substituant. S'il y en a plusieurs, chacun est précédé de N et ils sont énoncés dans l'ordre alphabétique.
|
|
|
|
N-éthyl-N-Méthylpropanamide |
N, N-Diméthylbutanamide |
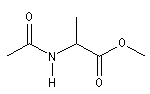
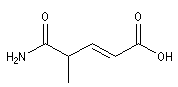
Lorsque la fonction amide n'est pas prioritaire elle est nommée par un préfixe. Le nom du groupe R-CO-NR' dérive de celui de R-CO-NHR' par remplacement de la terminaison amide par amido.
Les amides qui comportent un groupe phényle sur l'atome d'azote sont nommés en utilisant la terminaison anilide.
|
|
|
|
2-Ethanamidopropanoate de méthyle |
Acide 4-carbamoylpent-2-énoïque |
- Série cyclique
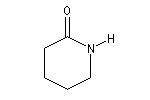
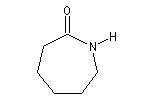
les amides cycliques sont appelés lactames.
|
|
 |
|
Azacyclohexan-2-one
(d-Valérolactame ) |
Azacycloheptan-2-one
(e-Caprolactame ) |
Imides
Le groupe fonctionnel d'un imide comporte deux groupes acyles liés au même atome d'azote. Ce sont donc des amides secondaires. On peut les considérer comme les analogues azotés des anhydrides.
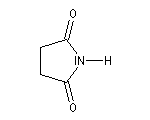
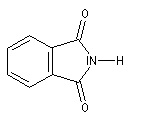
Les termes les plus importants sont les imides cycliques comme le succinimide ou le phtalimide.
|
|
 |
|
Butanimide
(Succinimide) |
Benzène-1, 2-dicarboximide
(Phtalimide) |
Etat naturel
Protéines
L'enchaînement d'aminoacides par des ponts amides (liaison peptidique) constitue la structure primaire des peptides et des protéines. La structure primaire de l'insuline a été élucidée en 1955 grâce à une méthode mise au point par le chimiste britannique Frederick Sanger. Ses travaux ont été couronnés par l'attribution du prix Nobel de chimie en 1958 [36]. F. Sanger est l'un des rares chimistes à avoir obtenu deux prix Nobel. En 1980,
il a reçu un second prix avec Paul Berg et Walter Gilbert pour leur contrbution à la détermination de la structure de l'ADN [37].
La structure secondaire de la myoglobine a été déterminée grâce à la diffraction des rayons X par M. F. Perutz et J. C. Kendrew de l'université de Cambridge. Ces derniers ont obtenu le prix Nobel en 1962 [38].
Antibiotiques possédant un cycle b-lactame
La pénicilline naturelle a été découverte en 1928 par le médecin anglais A. Fleming à partir de souches du champignon microscopique penicillium notatum. Ce n'est qu'en 1940 que deux biochimistes H. W. Florey et E. B. Chain réussirent à isoler et à purifier
la pénicilline. Ce travail leur a valu le prix Nobel de médecine en 1945 conjointement avec Fleming [28]. La structure de la pénicilline a été établie par la chimiste et cristallographe britannique Dorothy Crowfoot Hodgkin en utilisant la diffraction des rayons X. Elle reçut le prix Nobel de chimie en 1964 pour avoir élucidé la structure de la pénicilline et celle de la vitamine B-12 [45].
|
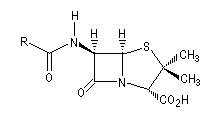
|
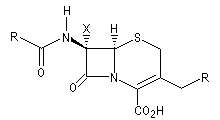
|
|
Pénicilline |
Céphalosporine |
Les b-lactames bloquent la synthèse de la paroi des bactéries.
Ce sont des inhibiteurs de la transpeptidase. Ils miment la structure tridimensionnelle de la séquence D-Ala-D-Ala. Cette enzyme, réagit avec la molécule antibiotique pour donner un complexe enzyme-produit fortement lié. L'enzyme catalyse la transformation de l'antibiotique pour donner un composé qui se lie à elle de façon irréversible.
Il s'agit d'un mécanisme qualifié d'inhibition-suicide.
Des méthodes permettant la synthèse totale des b-lactames ont été mises au point. Une des difficultés liée à la préparation du cycle b-lactame, est sa grande fragilité et son extrême sensibilité à l'hydrolyse. Le chimiste américain R. B. Woodward a achevé la synthèse de la céphalosporine C en 1966. La conférence qu'il prononça lors de la réception de son prix Nobel est consacrée pour l'essentiel à ce travail [21], [25].
Parmi les différentes voies d'accès aux b-lactames, la réaction de Staudinger utilise la cycloaddition [2+2] entre une imine et un cétène. Il existe une version diastéréosélective de cette réaction.
Beaucoup d'antibiotiques sont fabriqués industriellement par hémisynthèse à partir d'un substrat commun, l'acide 6-aminopénicillanique obtenu à partir du champignon penicillium chrysogenum.
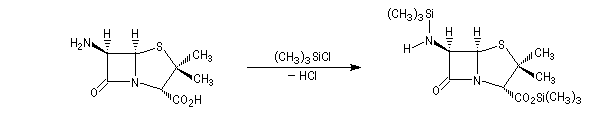
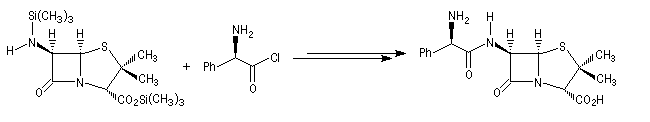
Macrolactames
Il existe de nombreux composés naturels comportant une structure de macrolactame. Certains possèdent des effets physiologiques très
importants et sont utilisés dans un but thérapeutique. Des exemples sont fournis par l'antiinflammatoire ascomycine [27], l'immunosuppresseur FK-506 ou la rapamycine [39].
|
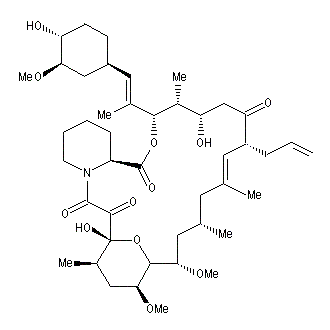
|
FK 506 est le nom d'un agent immunosupresseur utilisé comme médicament anti-rejet après une greffe d'organe. Ses effets sont de dix à cent fois supérieurs à ceux de la Cyclosporine. De nos jours, ce composé est commercialisé sous les noms : Tacrolimus, ProGraf ou Fujimycine. Il a été découvert en 1984 au Japon par des chercheurs de la firme pharmaceutique Fujisawa puis isolé à partir de champignons microscopiques Streptomyces tsukubaensis.
La molécule comporte 14 centres chiraux. Sa synthèse a été effectuée par des chercheurs de Merck en 1989 [63]. FK-506 est au coeur du livre de Barry Werth paru en 1994 intitulé "The Billion-dollar Molecule" qui raconte la création et le développement de la firme pharmaceutique américaine
Vertex. |
La synthèse totale de ce type de composés a surtout été développée à partir des années 90. Un exemple de réaction de macrolactamisation est présenté dans le chapitre relatif aux dérivés d'acides [64].
Quelques amides et imides importants
|
|
Une méthode de préparation du DMF comme celle de beaucoup d'amines substituées à l'azote, consiste à réaliser la réaction d'aminolyse d'un ester.
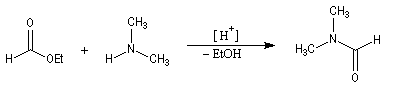 Le DMF est un solvant dipolaire aprotique très utilisé en chimie organique. Il est utilisé comme réactif dans la réaction de formylation de Vilsmeier-Haack
Le DMF est un solvant dipolaire aprotique très utilisé en chimie organique. Il est utilisé comme réactif dans la réaction de formylation de Vilsmeier-Haack
|
|
|
A la température ordinaire, l'éthanamide se présente sous la forme de cristaux incolores déliquescents. Une méthode de préparation découverte par Hofmann, consiste à chauffer le
sel d'ammonium de l'acide éthanoïque.
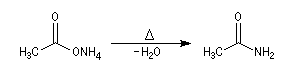
Une déshydratation plus poussée conduit à l'éthanenitrile.
|
|
|
Le e-caprolactame peut être préparé par réaction entre la cyclohexanone et le sulfate d’hydroxylamine ce qui fournit la cyclohexanone-oxime. En présence d'acide sulfurique concentré celle-ci fournit le caprolactame à la suite d'une transposition de Beckmann.
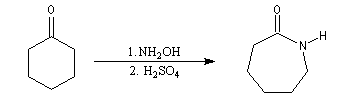
|
Une autre synthèse du caprolactame consiste a préparer préparer la caprolactone par oxydation de Baeyer-Villiger de la cyclohexanone. La caprolactone est ensuite traitée par l'ammoniac.
La polymérisation par ouverture du cycle du e-caprolactame founit le nylon 6 ou perlon. Ce polyamide possède des propriétés voisines
de celles du Nylon 6-6. Sa synthèse a surtout permis aux entreprises concurrentes de du Pont de Nemours de pénétrer le marché des polyamides à une époque où le
brevet du Nylon 6-6 était déposé [31], [32].
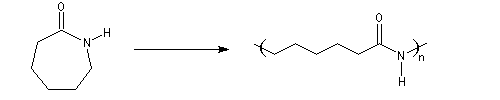
|
|
Le succinimide peut être obtenu par réaction entre l'anhydride succinique et l'ammoniac.
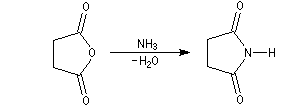
|
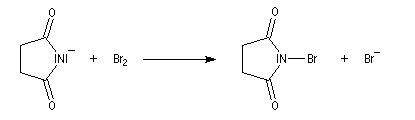
Le N-bromosuccinimide (NBS) est préparé par réaction entre le dibrome et l'ion imidate obtenu en déprotonant le succinimide.
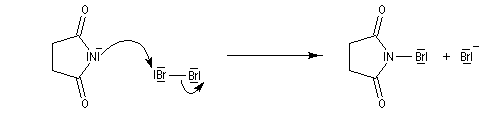
Le NBS est utilisé dans plusieurs réactions importantes. On peut citer :
Le N-chlorosuccinimide (NBS) est également un réactif utile utilisé notamment dans l'oxydation de Corey-Kim. des alcools.
Les N-méthoxy, N-méthylamides ou amides de Weinreb du nom du chimiste américain S. M. Weinreb qui les a introduits dans les années 80 sont des intermédiaires de synthèse importants en chimie organique moderne [65].
On les prépare à partir de nombreux substrats notamment les acides et les dérivés d'acides. Une méthode consiste à faire réagir un chlorure d'acyle avec la N, O-diméthylhydroxylamine (DMHA) [46].
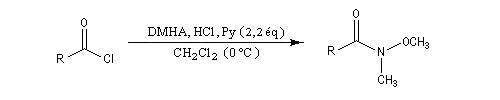
Les amides de Weinreb substitués par des a-aminoacides sont des réactifs particulièrement intéressants pour la synthèse stéréosélective de molécules chirales. Plusieurs sont disponibles commercialement.
Leur réduction par LiAlH4 ou le DIBAL-H permet la préparation d'aldéhydes. Par réaction avec un composé organométallique on obtient des cétones.
Propriétés physiques
Constantes physiques
|
Composé |
M (g.mol-1) |
TF (°C, 1 bar) |
(20 °C, 1 bar) |
|
HCONH2 |
45 |
2,5 |
liq |
|
HCON(CH3 )2 |
73 |
_ |
liq |
|
CH3CONH2 |
63 |
81 |
sol |
Structure électronique
La paire non liante sur l'atome d'azote est délocalisée sur l'ensemble des trois atomes de la fonction. Dans la méthode de la résonance, cela se traduit par l'existence de
deux formes limites. La forme dipolaire acquiert ici une certaine importance car :
- l'atome d'oxygène plus électronégatif que l'atome d'azote draîne la densité alectronique ;
- l'azote moins électronégatif que l'oxygène est davantage donneur par effet +M que ce dernier.
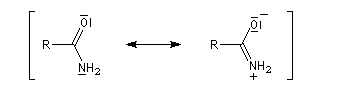
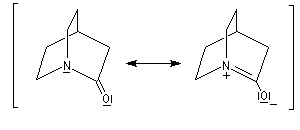
Un amide bicyclique tel que celui représenté ci-dessous est moins stable qu'un amide ordinaire. La résonance est empêchée du fait de l'impossibilité d'avoir une double liaison en tête de pont (règle de Bredt).
En résumé, l'atome de carbone d'un amide est le moins électrophile de tous les dérivés d'acides. Les amides ont une réactivité électrophile moindre que celle des
esters.
Spectroscopie
Spectroscopie infrarouge
Les valeurs données ci-dessous sont des valeurs moyennes.
|
s (cm-1 ) |
1425 |
1680 |
3250 |
|
Vibration |
élongation C-N |
élongation C=O |
élongation N-H |
On notera que le nombre d'onde de la vibration d'élongation du groupe carbonyle est inférieur à celui des cétones. On l'interprète comme un effet de
la conjugaison.
Le nombre d'onde de la vibration d'élogation N-H est modifié par la présence de liaisons H.
La spectroscopie est un moyen de distinguer les amides primaires secondaires et tertiaires.
- amides non substituées : deux bandes par exemple 3350 cm-1 (symétrique) et 3170 cm-1 (asymétrique) ;
- amides N-substituées : une seule bande 3210 cm-1 (N–éthylpropanamide) ;
- amides N,N-disubstituées : pas de bande.
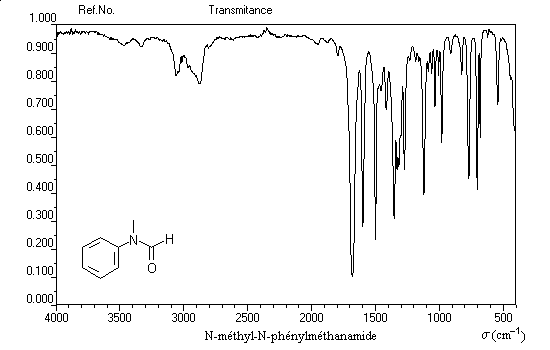
On notera que le nombre d'onde des vibrations des liaisons N-H associées par liaison hydrogène subissent un abaissement compris 20 cm-1 et 30 cm-1 par rapport aux liaisons non associées.
Spectroscopie de RMN
Sur le spectre de RMN du N, N-diméthylformamide, on observe que le signal des groupes méthyles se présente sous la forme d'un doublet pour un déplacement chimique légèrement inférieur à d = 3 ppm.
Cela signifie que la rotation autour de la liaison C-N n'est pas parfaitement libre. Plus précisément, à la température ordinaire :
- le groupe en cis par rapport à l'oxygène sort à 2,97 ppm ;
- le groupe en trans sort à 2,88 ppm.
Lorsque la température augmente, les deux signaux se fondent en un seul à la température de coalescence de 111 °C.
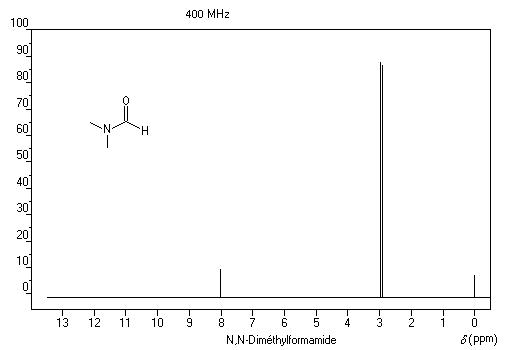
Cette restriction de la rotation trouve son origine dans la délocalisation du doublet de l'azote qui confère à la liaison C-N un caractère partiel de liaison double.
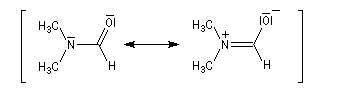
La barrière énergétique qui sépare les deux niveaux énergétiques est de l'ordre de 80 kJ.mol-1.
Propriétés acido-basiques
Basicité
Les amides sont des bases très faibles, beaucoup plus faible que les amines. Cela peut s'expliquer par le fait que le doublet de l'atome d'azote est délocalisé sur plusieurs atomes. La protonation s'effectue de préférence au niveau de
l'atome d'oxygène. La protonation sur l'atome d'azote détruit la conjugaison.
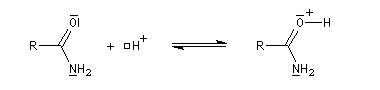
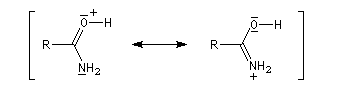
Tautomérie iminol-amide
Chez les amides qui possèdent au moins un atome d'hydrogène sur l'atome d'azote, la mobilité protonique se manifeste par la tautomérie amide-iminol. L'équilibre est en faveur de l'amide, thermodynamiquement plus stable que l'iminol du fait de la grande énergie de liaison du carbonyle et de la conjugaison entre le doublet non liant de l'azote et le carbonyle dans l'amide formé.
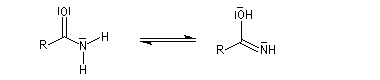
Le mécanisme est du même type que celui de la tautomérie céto-énolique.
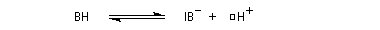
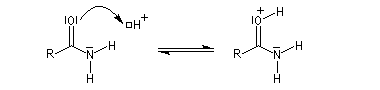
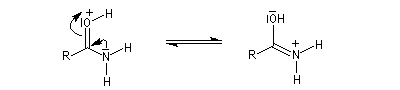
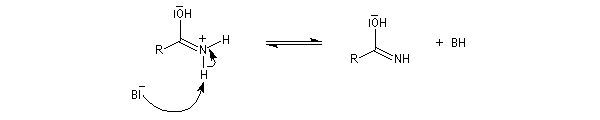
Acidité
Les amides sont des acides très faibles (pK ~ 16), mais notablement plus acides que l'ammoniac (pK = 33). L'anion amidate est stabilisé par résonance.
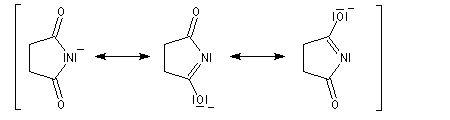
Les imides (pK ~ 8,5) sont beaucoup plus acides que les amides ordinaires (pK ~ 16).
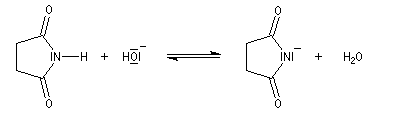
Ce résultat s'explique par le fait que la base conjuguée d'un imide est stabilisée par résonance.
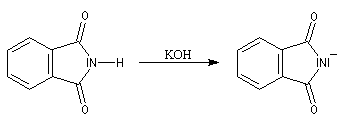
La bromation de l'ion imidate permet la préparation du N-bromosuccinimide (NBS).
L'alkylation des ions imidates est à la base de la synthèse de Gabriel des amines primaires.
Déshydratation des amides
Les amides conduisent aux nitriles sous l'action d'agents déshydratants comme PCl5, P4O10, SOCl2. Cette déshydratation constitue une manifestation du caractère labile de la liaison N-H.
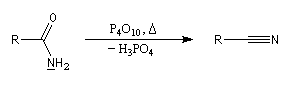
Avec PCl5, le mécanisme simplifié est le suivant.
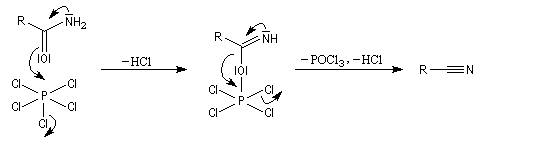
Inversement, l'hydratation d'un nitrile fournit l'amide. La plupart du temps, la réaction se poursuit pour donner l'acide carboxylique ou l'ion carboxylate correspondant.
Réactions de réduction
Réduction en aldéhyde
Les amides sont réduits en aldéhydes par le bis (2-méthylpropyl) aluminium Al(i-Bu)2H (diisobutylaluminium : DIBAL-H) un hydrure neutre à - 78 °C.
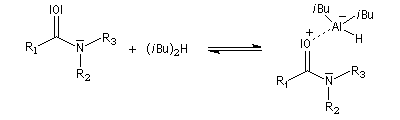
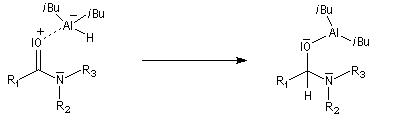
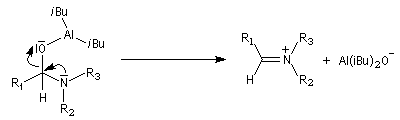
L'ion immonium est ensuite hydrolysé en aldéhyde.
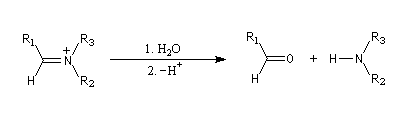
Voici des exemples de ce type de réaction.
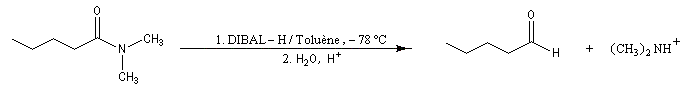
La réduction des amides de Weinreb est très utile dans les synthèses stéréosélectives car le centre chiral en alpha de la fonction amide est stable à la température de travail.
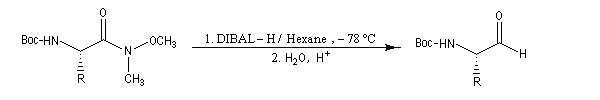
Ces amides peuvent également être réduits en aldéhyde par LiAlH4 dans certains cas. Dans l'exemple suivant, le produit (aldéhyde de Garner) est obtenu avec un excellent rendement. Cet aldéhyde est un substrat très utile dans la synthèse stéréosélective des sucres [47].
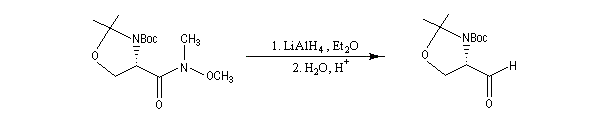
Réduction en amine
Lorsqu'on opère avec LiAlH4, la réaction va généralement jusqu'au stade de l'amine. Car l'ion immonium formé est réduit.
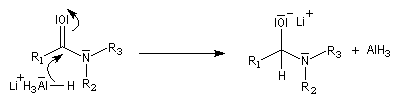
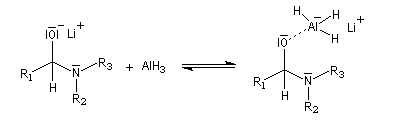
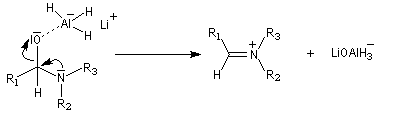
L'ion immonium est ensuite réduit en amine sous l'action de LiAlH4
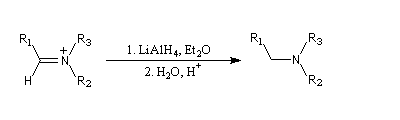
Voici un exemple de cette réaction.
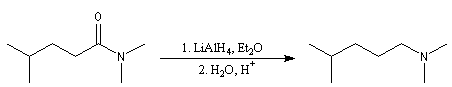
Réactions d'addition des organométalliques
Organomagnésiens et organolithiens
Le produit d'addition d'un organomagnésien sur un amide est thermodynamiquement instable par rapport au composé carbonylé du fait de la grande énergie de liaison du carbonyle. Cet intermédiaire est cinétiquement inerte car NR2-
est un très mauvais nucléofuge ce qui peut être mis en relation avec la basicité très forte des ions amidures.
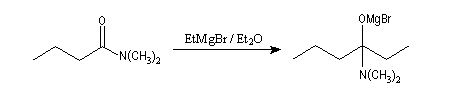
Le passage en milieu acide entraîne l'élimination de R2
NH2+ à partir de l'intermédiaire. Ce nucléofuge est en effet bien meilleur que NR2- car c'est une base beaucoup plus faible. On obtient une cétone.
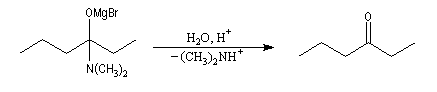
Il est impératif d'utiliser un amide N, N-disubstitué dans cette réaction. Avec un amide non substitué, le magnésien arrache un atome d'hydrogène à l'amide du fait de sa basicité. Il est alors détruit.
Réactions avec les amides de Weinreb
Les N-méthoxy, N-méthylamides ou amides de Weinreb sont des substrats qui réagissent avec les organomagnésiens et les organolithiens dans des conditions douces pour donner des cétones. La réaction fait intervenir un intermédiaire dans lequel le métal est complexé par les atomes d'oxygène. Il s'agit d'un exemple de complexe chélaté [65]. Le complexe est suffisamment stable pour ne pas se décomposer dans le milieu réactionnel à la différence de ce qui se passe avec un chlorure d'acyle ou un ester.
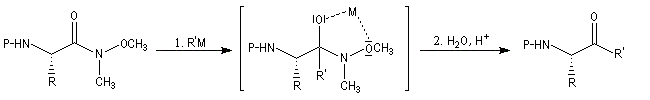
La cétone est libérée par hydrolyse.
Voici un exemple d'une telle réaction. Le symbole -Boc représente le groupe butoxycarbonyle, qui sert de groupe protecteur de la fonction amine. Lorsque la réaction est effectuée à basse température (- 78 °C)
la configuration absolue du centre chiral du substrat est conservée.

Application à la synthèse des b-cétophosphonates
L'addition d'un organolithien convenable sur un amide de Weinreb permet de préparer des dialkylesters de l'acide b-cétophosphonique utilisés dans la réaction de Wittig-Horner.
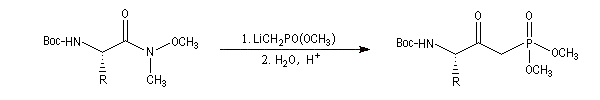
Hydrolyse des amides
Résultats expérimentaux
L'hydrolyse des amides possède des points communs avec l'hydrolyse des esters. Mais les conditions expérimentales sont plus sévères. En effet, par rapport aux esters, les amides possèdent :
- un atome de carbone moins électrophile ;
- un mauvais nucléofuge : l'ion amidure.
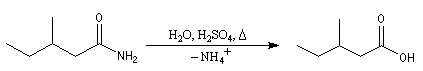
Du fait de leur relative inertie chimique, les amides sont fréquement employés comme groupe protecteur de la fonction amine.
Hydrolyse en milieu basique
La réaction débute par l'attaque du carbonyle de l'amide par l'ion hydroxyde.
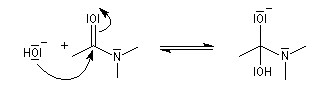
La réaction est lente car les amidures sont de mauvais nucléofuges à corréler avec une basicité élevée.
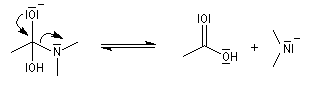
Les ions amidures étant des bases très fortes, la dernière étape est irréversible.
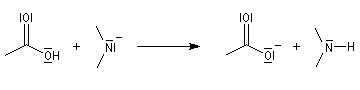
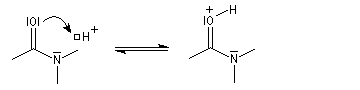
Hydrolyse en milieu acide
Le carbonyle est protoné ce qui a pour effet d'exalter le caractère électrophile du carbone. Il s'agit d'un exemple d'assistance électrophile.
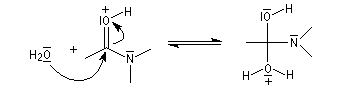
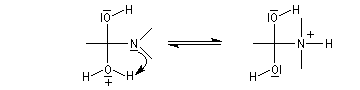
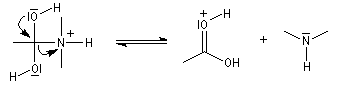
L'acide protoné est un acide beaucoup plus fort que l'ion ammonium. La dernière étape est irréversible.
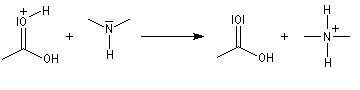
Notons que l'ion H+ n'est pas, à proprement parler, un catalyseur puisqu'il est consommé dans la dernière étape.
Réactions avec les alcools
L'alcoolyse des amides conduit aux esters. La réaction rappelle celle de transestérification mais la température doit être plus élevée. La réaction est lente même en présence d'un catalyseur.
Synthèse des amines primaires de Gabriel
L'un des problèmes soulevé par la réaction d'alkylation d'Hofmann est la suralkylation de l'amine formée.
Le chimiste allemand S. Gabriel a mis au point une synthèse des amines primaires basée sur l'alkylation d'un imide dérivé de l'acide phtalique [60]. La réaction s'effectue en plusieurs étapes :
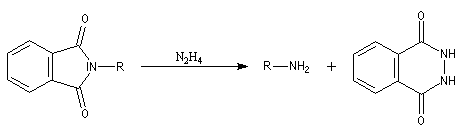
Un exemple d'application de la synthèse de Gabriel est la préparation de la benzylamine qu'on trouvera à la référence [6].
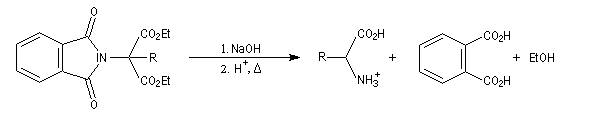
Lorsque le dérivé halogéné est le malonate de diéthyle, la réaction permet une intéressante synthèse d'aminoacide.
- On commence par effectuer l'alkylation de l'ion imidate par le dérivé halogéné ;
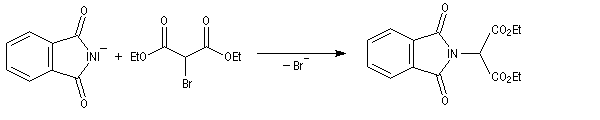
- le composé obtenu porte un atome d'hydrogène acide qui est facilement enlevé par une base ;
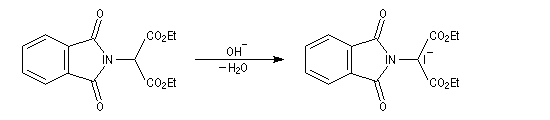
- l'alkylation peut alors être réalisée au moyen d'un dérivé halogéné ;
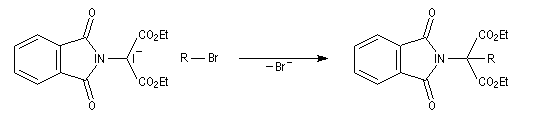
- il ne reste plus qu'à hydrolyser l'imide substitué pour obtenir l'aminoacide souhaité.
Propriétés des atomes d'hydrogène portés par l'atome de carbone en a
Enolates d'amides
L'hydrogène en a de la fonction amide est acide. Le pK du couple (amide/énolate) étant de l'ordre de 30, il peut être arraché par une base très forte comme le LDA. Les amides non substitués à l'azote fournissant dans ces conditions l'anion amidate, on utilise des amides N, N-disubstitués dans ce type de réaction.
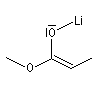
Formation d'énolates Z et E
L'obtention d'énolates Z et E à partir de composés carbonylés a été vue dans le chapitre correspondant. Compte-tenu de la méthode utilisée pour définir
les stéréodescripteurs Z et E, il faut faire attention qu'à une double liaison avec des substituants O-Li et alkyle en cis dans un énolate d'ester, correspond à une configuration relative E, alors que la même
relation cis autour de la double liaison d'un énolate d'amide, correspond à une configuration Z.
|
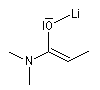
|
|
|
Enolate d'amide (Z) |
Enolate d'ester (E) |
Avec le LDA dans le THF, la formation de l'énolate Z est favorisée.
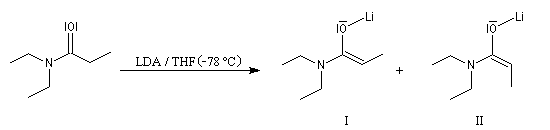
Alkylation des énolates d'amides
Les amides dérivés d'oxazolidinones permettent la préparation d'énolates Z avec une grande stéréosélectivité. Ceux-ci peuvent être alkylés par un dérivé halogéné de façon diastéréosélective selon la méthode de D. Evans. Après séparation chromatographique des diastéréoisomères, l'auxilliaire est détaché. Par hydrolyse on obtient des acides et par réduction des alcools. Dans les deux cas, les excès énantiomériques sont excellents.
Réactions propres aux amides
Réaction avec l'acide nitreux
La nitrosation est la réaction entre un amide et l'acide nitreux HNO2
. Ce dernier est un composé instable vis à vis de sa dismutation en NO3- et NO.
Il est préparé dans le milieu réactionnel en acidifiant une solution de nitrite de sodium par l'acide chlorhydrique.
La formation de l'électrophile a été vue dans le chapitre consacré à la nitrosation des amines.
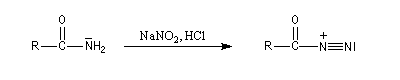
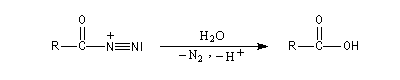
Dégradation d'Hofmann
Cette réaction découverte par Hofmann à la fin du dix neuvième siècle, permet de passer d'un amide à une amine primaire possédant un atome de carbone de moins [62]. Le mécanisme est détaillé ci-dessous.
La base arrache un proton à l'amide.
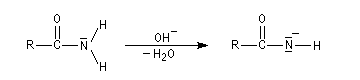
L'ion amidate formé réagit avec le dibrome pour donner une N-bromamide.
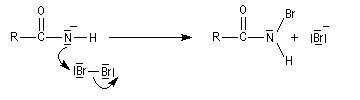
La base arrache un proton de la bromamide pour former un anion.
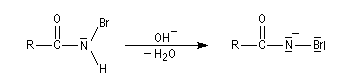
L'anion perd un ion bromure pour former un nitrène.
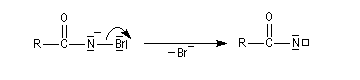
Le nitrène subit une transposition en isocyanate. Celle-ci rappelle la transposition de Wolff qui conduit aux cétènes.
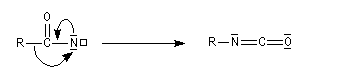
La réaction entre l'ion hydroxyde et l'isocyanate fournit un carbamate.
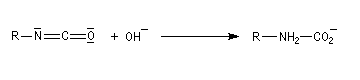
Après élimitation de CO2 à partir du carbamate, on obtient une amine.
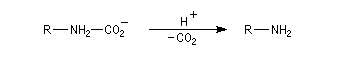
Un exemple d'application est la préparation de la cyclobutylamine [29].
Bibliographie
Ouvrages expérimentaux
[1] Manuel d'expériences de chimie - UNESCO Société chimique de France - Université de Montpellier.
[2] P. Rendle, M. V. Vokins, P. Davis, Experimental Chemistry (Edward Arnold).
[3] L. F. Fieser, K. L. Williamson - Organic Experiments (D. C. Heath and Company).
[4] R. Adams, J. R. Johnson, C. F. Wilcox - Laboratory Experiments in Organic Chemistry (The Macmillan Company, Collier-Macmillan Limited).
[5] G. K. Helmkamp, H. W. Johnson Jr, Selected Experiments in Organic Chemistry (W. H. Freeman and Co).
[6] Vogel's Textbook of Practical Organic Chemistry (Longman).
[7] J. R. Mohrig, D. C Neckers, Laboratory Experiments in Organic Chemistry (D. Van Nostrand Company).
[8] R. Q. Brewster, C. A. Van der Werf, W. E. Mc Ewen, Unitized Experiments in Organic Chemistry (D. Van Nostrand Company).
[9] F. G. Mann, B. C. Saunders, Practical Organic Chemistry (Longman).
[10] M. T. Yip, D. R. Dalton, Organic Chemistry in the Laboratory (D. Van Nostrand Company).
[11] Miller, Neuzil, Modern Experimental Organic Chemistry (DC Heath, 1982)
[12] R. K. Slingh, S. Danishefsky, Org. Synth. 60, p. 66.
Ouvrages théoriques
[13] A. Streitwieser Jr, C. H. Heathcock - Introduction to Organic Chemistry, Macmillan Publishing Co.
[14] J. March - Advanced organic chemistry, Wiley Interscience.
[15] F. A. Carey, R. S. Sunberg - Advanced Organic Chemistry, Plenum Press 1990.
[16] N. T. Ahn - Introduction à la chimie moléculaire, Ellipses 1994.
[17] R. Brückner - Mécanismes réactionnels en chimie organique, De Boeck Université 1999.
Liens
[20] Classification des amides (IUPAC)
[21] R. B. Woodward - The Nobel Prize in chemistry 1965
[25] R. B. Woodward speaking on cephalosphorin C
[26] R. B. Woodward by James B. Hendrickson, who is Professor of Chemistry at Brandeis University, Waltham, Massachusetts
[27] Ascomycins Macrolactams
[28] Sir Alexander Fleming The Nobel Prize in Physiology or Medicine
[29] Cyclobutylamine from cyclobutanecarboxamide by Merrick R. Almond, Julie B. Stimmel, E. Alan Thompson, and G. Marc Loudon.
[30] Intermediates in the Ing-Manske reaction
[31] Propriétés du Nylon 6
[32] Synthesis of Nylon 6
[33] Antibiotiques
[34] Tacrolimus
[35] Insulin : structure of a protein hormone
[36] F. Sanger The Nobel Prize in Chemistry 1958
[37] P. Berg, W. Gilbert, F. Sanger The Nobel Prize in Chemistry 1980
[38] M. F. Perutz, J. C. Kendrew The Nobel Prize in Chemistry 1962
[39] Total synthesis of Rapamycin
[40] Macrolacatamization
[41] Réduction des amides par les boranes
[42] Hofmann degradation
[43] Amines as bases
[44] Assymetric alkylation of enolates
[45] Dorothy Crowfoot Hodgkin The Nobel Prize in Chemistry 1964
[46] Amides de Weinreb
[47] Garner aldehyde by Xifu Liang, Jens Andersch and Mikael Bols
Articles
[60] Gabriel, S. Ber. 20, 2224 (1887).
[61] Ing, H.R.; Manske, R.H.F. J.Chem.Soc.,1926 , 2349.
[62] Hofmann, A.W. Ber., 1881, 14, 2725.
[63] Jones, T.K., Mills, S.G., Reamer, R., Askin, D., Desmond, R., Volante, R.P. and Shinkai, I. Journal of the American Chemical Society 111, 1157-1159 (1989)
[64] Lactamization of 11-Azidoundecanoic Acid Derivatives – DMAP as Catalyst by I. Bosch, P. Romea, F. Urpi, and J. Vilarrasa, Tetrahedron Lett., 34, 4671 (1993)
[65] Nahm, S.; Weinreb, SM Tetrahedron Lett. 1981, 22, 3815-3818
RETOUR AU MENU DU COURS
Vous pouvez, si vous le souhaitez, utiliser le contenu de cette page dans un but pédagogique et non commercial.
Texte, dessins, photographies : Gérard Dupuis - Lycée Faidherbe de LILLE
décembre 2010