|
|
48èmes Journées Nationales de l'Union des Physiciens Atelier A 6 : Molécules & Internet
Gérard Dupuis - Lycée Faidherbe de Lille
|
|
|
48èmes Journées Nationales de l'Union des Physiciens Atelier A 6 : Molécules & Internet
Gérard Dupuis - Lycée Faidherbe de Lille
|
Visualisation des molécules
La visualisation en trois dimensions de molécules constitue une partie importante de la stéréochimie. Depuis maintenant près de deux siècles, les chimistes à la suite des cristallographes (dans ce domaine R.J. Haüy fut un précurseur) ont rivalisé d'imagination afin de représenter molécules et cristaux pour les besoins de l'enseignement et de la recherche. Pour juger de l'importance que les chimistes ont toujours accordé à la modélisation rappelons que c'est en partie grâce à des modèles construits avec du fil de fer, que J. Watson et F. Crick ont eu l'intuition de la structure en double hélice de l'ADN.
Les progrès accomplis en informatique depuis une vingtaine d'années permettent à tout chimiste de visualiser des molécules en s'aidant d'un micro-ordinateur grâce à des programmes dont certains appartiennent au domaine public ou sont libres d'utilisation. Loin de se limiter à une simple représentation statique, ces programmes permettent de déplacer la molécule en trois dimensions et d'afficher les principaux paramètres moléculaires : distances interatomiques, angles entre les liaisons, rayons de Van der Waals etc.
De nombreuses banques de données réparties à travers le monde, proposent des fichiers décrivant les caractéristiques de molécules inorganiques et organiques. Qu'ils représentent des molécules très simples ou comportant plusieurs centaines d'atomes, ces fichiers sont facilement accessibles via le réseau Internet. Après avoir été téléchargés, ils sont utilisables en ligne grâce à un plug in associé au navigateur ou enregistrés sur un support afin d'être visualisés hors connexion.
Atelier : molécules et internet
L'étude complète des fonctionnalités du programme CHIME est assez longue et risque d'être rapidement fastidieuse. Aussi, après avoir appris comment lancer CHIME et mémorisé les commandes souris, vous avez intérêt à passer rapidement à l'étude de différents exemples. Vous pourrez ensuite, lorsque le besoin se présentera, revenir à l'étude plus détaillée des autres commandes.
Remerciements :
Historique
En 1989 Roger Sayle élève de l'Imperial College de Londres, cherche à écrire un programme qui permette la visualisation et le déplacement de molécules en trois dimensions. Il crée la première version du programme RasMol (de Raster Molecules). Devenu étudiant à l'université d'Edinbourg, il continue de développer son programme en collaboration avec un cristallographe de cette université : le Dr. Andrew Coulson. En 1993 après avoir obtenu son Ph. D, R. Sayle décide généreusement de placer le code source de RasMol dans le domaine public. Depuis cette date, ce programme est à la libre disposition de la communauté scientifique internationale. Plusieurs auteurs ont poursuivi le travail de R. Sayle et il existe actuellement de nombreuses versions du programme initial. RasMol a été conçu à l'origine pour la visualisation de grosses molécules (protéines, acides nucléiques, enzymes) mais il permet naturellement celle de molécules plus petites.
La création de ce programme est décrite dans : une courte histoire de RasMol par son auteur, R. Sayle.Les différentes versions de RasMol
Elle peut être téléchargée sur le site de Biogeo où l'on trouvera d'intéressantes précisions concernant sa mise en œuvre.
E. Martz (UMass) a créé une liste commentée de commandes utilisables par RasMol.
Origine
T. Maffet et B. van Vliet de la firme MDL Informations Systems, Inc ont utilisé 16 000 lignes du code source de RasMol pour créer et développer le programme CHIME 1.0 (CHemical MIME) en 1997. F. Adler et J. Holt ont achevé la version 2.0 en 1998. Ce programme est libre d'utilisation et peut être téléchargé gratuitement sur le site de MDLI. A la différence de RasMol, CHIME n'appartient pas au domaine public et son code source est la propriété de MDLI.
CHIME possède certaines fonctionnalités de RasMol mais il ne fonctionne pas de façon autonome. C'est un module externe (plug-in) qui se greffe sur un navigateur. Ce qui suit concerne la version 2.0.3. (1999).
Hardware :
|
Processeur |
Mémoire vive |
Ecran |
|
80486 minimum, 90 MHz ou Pentium recommandé |
16 Mo RAM minimum, 32 Mo ou plus recommandés |
VGA 800 ´ 600 minimum |
Logiciels :
CHIME a été conçu à l'origine pour être greffé sur Navigator de Netscape.|
Système d'exploitation |
Navigateur |
|
Win 95, Win 98 Win NT 4.0 |
Navigator v 3.0x et Communicator v 4.x de Netscape |
Il existe une version pour Mac disponible sur le site de MDLI.
Ce qui suit est relatif aux systèmes d'exploitation Windows 95, Windows 98, Windows NT 4.0. L'installation est effectuée par téléchargement sur le site de MDL. Les fichiers sont automatiquement placés dans deux sous-répertoires du dossier navigateur IE ou Netscape :
Sous-répertoire Chime
Sous-répertoire Plugins
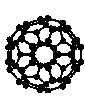
La molécule de C-60
|
|
L'image constitue l'extrémité d'un lien hypertexte dont la cible est le fichier c-60.pdb. Vérifier que son adresse
est affichée dans la barre d'état en bas du navigateur lorsqu'on place le pointeur sur l'image. Si elle n'est
pas visible, aller dans le menu affichage du navigateur et sélectionner barre d'état. Vous pouvez lancer CHIME :
|
|
Action |
Commande |
|
ouvrir un menu déroulant |
bouton droit |
|
rotation X,Y |
bouton gauche |
|
rotation Z |
shift-bouton droit |
|
déplacer la molécule dans la fenêtre |
ctrl-bouton droit |
|
agrandir la molécule (zoom) |
shift-bouton gauche |
Menus déroulants
En cliquant sur le bouton droit, on fait apparaître des menus déroulants subdivisés en sous-menus. Nous ne passerons en revue que les principaux.
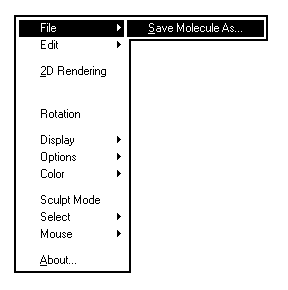
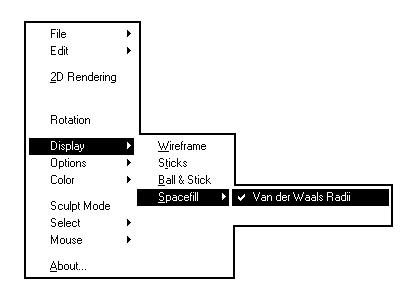
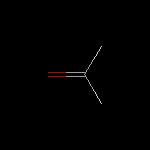
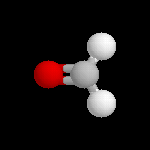
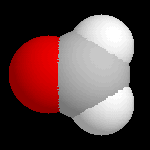
Les images ci-dessous représentent la molécule de méthanal en utilisant successivement ces quatre modes de visualisation.
|
|
|
|
|
|
Wireframe |
Sticks |
Ball & sticks |
Spacefill |
Permet de chosir la couleur avec laquelle sont représentés les atomes.
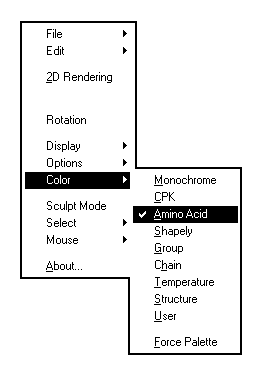
Les couleurs conventionnelles sont les mêmes que celles qui sont utilisées pour les modèles moléculaires ordinaires développés par Corey, Pauling et Kultun (système CPK). Elles sont données ci-dessous :
|
Elément |
Couleur |
Code RGB |
|
carbone |
gris clair |
[200, 200, 200] |
|
oxygène |
rouge |
[255, 0, 0] |
|
hydrogène |
blanc |
[255, 255, 255] |
|
azote |
bleu clair |
[143, 143, 255] |
|
soufre |
jaune |
[255, 200, 50] |
|
phosphore |
orange |
[255, 165, 0] |
|
chlore |
vert |
[0, 255, 0] |
|
brome, zinc |
marron |
[165, 42, 42] |
|
sodium |
bleu |
[0, 0, 255] |
|
fer |
violet |
[160, 32, 240] |
|
calcium, métaux |
gris foncé |
[128, 128, 144] |
|
inconnu |
rose profond |
[255, 20,147] |
|
Amino-acide |
Couleur |
Code |
|
ASP, GLU |
rouge brillant |
[230, 10,10] |
|
LYS, ARG |
bleu |
[20, 90, 255] |
|
PHE, TYR |
bleu moyen |
[50, 50, 170] |
|
GLY |
gris clair |
[235, 235, 235] |
|
ALA |
gris foncé |
[200, 200, 200] |
|
HIS |
bleu pâle |
[130, 130, 210] |
|
CYS, MET |
jaune |
[230, 230, 0] |
|
SER, THR |
orange |
[250, 150, 0] |
|
ASN, GLN |
cyan |
[0, 220, 220] |
|
LEU, VAL, ILE |
vert |
[15, 130, 15] |
|
TRP |
rose |
[180, 90, 180] |
|
PRO |
chair |
[220, 150, 130] |
Cette fonction permet d'assigner une couleur à chaque chaîne d'une protéine ou d'un acide nucléique. Cela permet
d'identifier plus facilement les différentes parties d'une structure. On peut ainsi, par exemple, mettre en évidence facilement la structure en double
hélice de l'ADN.
Cette fonction permet d'avoir une idée de la mobilité d'un atome dans une chaîne. Aux valeurs élevées correspondent des couleurs chaudes (rouge)
aux faibles valeurs, des couleurs froides (bleu).
Cette fonction permet de visualiser la structure secondaire des protéines. Les hélices a sont colorées en magenta, les feuillets
b sont colorés en jaune, les coudes b en bleu. Les autres résidus sont en blanc.
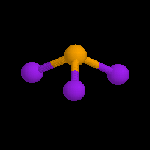
Illustration de la méthode VSEPR
Vous pouvez utiliser la même démarche pour afficher les caractéristiques géométriques
de molécules inorganiques simples afin d'illustrer la méthode VSEPR.
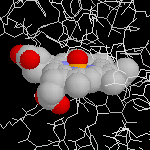
Phénol
L'image ci-dessous représente la molécule de Phénol.
|
|
|
Comme deuxième exemple vous pouvez observer la molécule d'adénosine triphosphate ATP et comparer les différentes représentations avec la formule dessinée en 2D.
Acides lactiques énantiomères
Visualiser l'acide (2R)-lactique dans une nouvelle fenêtre. Diminuer la taille de cette fenêtre en cliquant sur un des angles du cadre et en faisant
glisser la souris le long d'une diagonale. Recentrer au besoin l'image de la molécule en utilisant la combinaison de touches ctrl-bouton droit. Visualiser de la même manière l'acide(2S)-lactique dans une nouvelle fenêtre placée à côté de la précédente. Vérifier que les molécules précédentes sont des énantiomères.
Complément sur les acides lactiques énantiomères.
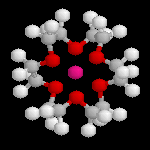
18-C-6
L'image ci-dessous représente un complexe entre l'ion K+ et l'éther-couronne 18-C-6. Ce composé a été synthétisé en 1967
par C. J. Pedersen qui a obtenu le prix Nobel de chimie en 1987.
|
|
|
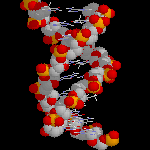
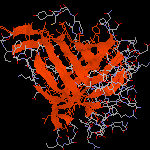
Molécule d'ADN
L'image ci-dessous représente un fragment d'une chaîne d'ADN. La structure en double hélice de cette molécule a été prévue par J. D. Watson (Harvard) et F. H. Crick (Cambridge) en 1953. Cette
structure a été pleinement confirmée au moyen de la diffraction des rayons X par R. Franklin et M. Wilkins (Londres), la même année. J. Watson et F. Crick ont obtenu le prix Nobel de médecine en 1960.
|
|
|
|
|
|
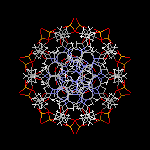
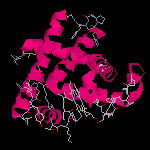
Molécule de myoglobine
L'image ci-dessous représente la molécule de myoglobine. Cette protéine permet la mise en réserve
du dioxygène dans les muscles. Sa structure a été déterminée grâce à la diffraction des rayons X
par M. F. Perutz et J. C. Kendrew de l'université de Cambridge. Ces derniers ont obtenu le prix Nobel en 1962.
|
|
|
|
|
|
|
|
|
|
|
|
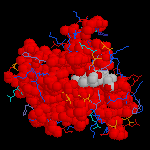
1RBP (Retinol Binding Protein)
L'image ci-dessous représente la molécule de 1RBP (retinol binding protein).
|
|
|
La nature du fichier qui est transféré est indiquée par la commande Content-type (type de contenu). Chaque type est constitué d'un type général qui indique la nature du fichier et d'un sous-type qui caractérise son format exact (exemple x-pdb). En 1994, H. S. Rzepa, P. Murray-Rust et B. J. Whitaker ont proposé la création d'un nouveau type MIME : le type chemical.
A chaque type/sous-type est associé une extension que le navigateur utilise pour repérer le fichier (exemple .pdb).
Lorsqu'on lance le navigateur celui-ci commence par inventorier les différents plug-in installés sur la machine et enregistre leur type MIME dans un répertoire. Lorsque le navigateur rencontre un fichier comportant l'extension .pdb il examine le répertoire contenant les types MIME et appelle le plug in adéquat.
Les formats reconnus par CHIME 2.0.3 sont mentionnés dans le fichier Npchime.txt. On les trouve également sur le site de MDL. Ce sont les suivants :
|
Description du type |
Type/sous-type MIME |
Extension |
Depuis la version |
|
MDL Molfile |
chemical/x-mdl-molfile |
.mol |
0.8 |
|
Brookhaven Protein Databank |
chemical/x-pdb |
.pdb |
0.8 |
|
IEMBL Nucleotide Format |
chemical/x-embl-dl-nucleotide |
.emb,.embl |
0.8 |
|
Minnesota Supercomputer Center's XMol XYZ |
chemical/x-xyz |
.xyz |
0.8 |
|
Gaussian Input File |
chemical/x-gaussian-input |
.gau |
0.8 |
|
Rasmol Script File |
application/x-spt |
.spt |
0.8 |
|
Mopac Input File |
chemical/x-mopac-input |
.mop |
0.8 |
|
Chemical Structure Markup Language File |
chemical/x-csml |
.csm,.csml |
0.8 |
|
MDL Transportable Graphics File |
chemical/x-mdl-tgf |
.tgf |
Future |
|
MDL RxnFile |
chemical/x-mdl-rxnfile |
.rxn |
1.0 |
|
JCAMP-DX File |
chemical/x-jcamp-dx |
.jdx,.dx |
2.0 |
|
RasMol Script File |
application/x-spt |
.spt |
0.8 |
|
RasMol Script File |
application/x-rasmol |
.scr |
2.0 |
On pourra se reporter à la page d'accueil du format MIME sur le site de l'Imperial College de Londres. Cette page propose des informations complémentaires sur les différents types et sous-types et constitue un point d'entrée vers de nombreuses ressources.
Il est possible de visualiser les types MIME reconnus et le cas échéant en ajouter d'autres. La procédure est la suivante :
Dans : description du type, on inscrit : Brookhaven Protein Databank
Dans : extension associée, on inscrit : pdb
Dans : Type de contenu (MIME), on inscrit : chemical/x-pdb
Compression de fichiers
CHIME reconnaît les fichiers compressés au moyen de GZIP. Des versions de GZIP (et GUNZIP pour décompresser) existent pour Windows, Mac, Unix etc.
Il faut cependant faire attention que les fichiers compressés sont sous forme binaire. Leur transfert en mode ASCII via un protocole ftp peut les corrompre.
Configuration
CHIME a été conçu à l'origine pour être greffé sur Navigator de Netscape. Il fonctionne avec MSIE 5.0 mais ce navigateur ne permet pas d'exploiter toutes ses fonctions.
Lors de l'utilisation de CHIME avec MSIE 5.0 on peut voir apparaître le message d'erreur suivant :
Chime fatal error ! an error occured inside the plug-in
Renderer Error : Unable to allocate frame buffer
Chime could not allocate enough memory to display molecule in 3D
Cela peut provenir d'une mauvaise configuration de MSIE 5.0. Pour y remédier :
Cela peut provenir d'une mauvaise définition des types de contenu (MIME) des fichiers. Il faut alors ajouter les ajouter comme cela a été indiqué précédemment.
Utilisation de scripts
Si le problème persiste il se peut que les pages visitées contiennent des scripts qui sont mal interprétés par MSIE 5.0. Il ne faut pas tenter de visiter ces pages avec IE 5.0 et se tourner vers un navigateur compatible. On pourra consulter
une liste
de navigateurs compatibles avec CHIME établie par E. Martz.
Recherche de molécules dans une banque de données
Micromolécules
Dans la plupart des banques de données regroupant des petites molécules, leurs noms sont présentés sous forme de liens hypertextes. On a alors le choix entre deux possibilités :
La liste de banques de données suivante n'est naturellement pas exhaustive. En naviguant dans ces banques vous pouvez facilement télécharger des fichiers de molécules variées sur le disque dur ou sur une disquette.
Macromolécules
Le nombre et la variété des macromolécules biologiques sont immenses. Il est essentiel de pouvoir effectuer une recherche aussi systématique que possible.
Utilisation de Chime dans une page web
Insertion d'une fenêtre graphique dans une page HTML
Comme on l'a déjà vu, l'utilisateur peut lancer CHIME en cliquant sur un lien hypertexte. Une molécule peut aussi être visualisée automatiquement dans une fenêtre graphique dont les dimensions ont été définies au préalable par le créateur de la page web.
L'insertion dans la page repose sur l'utilisation de la balise EMBED.
La ligne d'instructions suivante insère le fichier cycloh-c.pdb du dossier images dans une fenêtre carrée de 300 pixels de côté. La molécule sera présentée sous forme de bâtonnets.
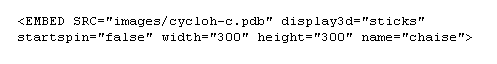
Cette procédure nécessite l'emploi d'un navigateur compatible tel que Communicator 4.7 de Netscape.
Ecriture de scripts
|
|
Construire ses propres molécules
Formats de fichiers
De nombreux formats de fichiers sont utilisés en modélisation moléculaire. En voici quelques-uns :
Une information complète sur les différents formats utilisables en modélisation moléculaire a été écrite par A. Dalby (J. Chem. Inf. Comput. Sci, 1992). Elle peut être téléchargée au format pdf (nécessite Acrobat Reader) sur le site de MDL. Notons qu'il est tout à fait possible de passer d'un format à un autre grâce à des programmes de conversion. Le programme BABEL permet la conversion entre les types précédents.
Mécanique moléculaire
L'énergie conformationnelle V d'une molécule peut être considérée comme la somme de plusieurs termes qui rendent-compte des interactions entre atomes liés et non-liés.
V = Vs + Vb + Vw + Vnb
V représente la somme des contraintes entre les atomes à l'intérieur de la molécule. C'est une fonction des coordonnées des atomes de la molécule. La mécanique moléculaire se fixe comme objectif la détermination de ces coordonnées en recherchant celles qui minimisent l'énergie totale par rapport à l'énergie d'une structure
de référence.
Plusieurs programmes informatiques basés sur les "champs de forces" MM1, MM2, MM3 etc. ont été développés dans les années 80, notamment par N. Allinger. Ils permettent le calcul d'un jeu de coordonnées qui minimisent V.
Plusieurs produits commerciaux permettent la construction de modéles moléculaires en 3D en mettant à profit cette méthode. En voici deux :
Le programme Sym-Apps 5.1 utilise le champ de forces MM2 pour construire une représentation en 3D d'une molécule à partir de la
connaissance de sa géométrie approximative.
On commence par dessiner la molécule en 2D au moyen d'un programme de dessin tel que ChemWindows qui est vendu par le même fabricant (Softshell). Ce dessin en 2D est copié dans le
presse-papier puis collé dans SymApps. Le calcul des coordonnées en 3D prend quelques secondes. Il reste alors à sauvegarder le fichier sous un format donné (.pdb, .mol ou autre). C'est en utilisant cette technique
qu'ont été construits la plupart des modèles utilisés dans le cours de chimie organique.
Les limites des programmes de modélisation
La méthode précédente est très utile pour créer des fichiers représentant des molécules complexes, mais elle n'est pas sans défauts. Prenons comme exemple la molécule
de ferrocène. Cette molécule a attiré l'attention des chimistes dès sa découverte en 1951 par L. Pauson. La structure
du ferrocène a été déterminée par R.B Woodward et G. Wilkinson : le fer est logé entre deux cycles cyclopentadiényles. Les programmes de modélisation basés sur la minimisation de l'énergie totale de la structure prédisent un angle entre les cycles de 36° qui correspond à
l'éloignement maximal des sommets des cycles. Des expériences de diffraction électronique et neutronique réalisées dans les années 80, ont montré que les deux cycles sont en réalité quasiment éclipsés puisque l'angle entre-eux
est voisin de 9°.
Pour décrire correctement la structure de la molécule on doit construire un fichier comportant les coordonnées des atomes calculées à partir des
valeurs expérimentales des distances interatomiques et des angles entre les liaisons. C'est cette méthode qu'a employée R.J Lancashire de l'Université West Indies pour construire le fichier .pdb du
ferrocène utilisé ici et qu'il a eu la gentillesse de me communiquer.
|
|
|
Complément sur la structure du ferrocène.
Par paramétrage direct des coordonnées C'est en utilisant cette méthode que les fichiers des molécules répertoriées dans la banque de données ont été construits, qui représentent les principales
géométries rencontrées dans l'enseignement de la méthode VSEPR.
Parmi les différents formats utilisables, le format .pdb est particulièrement avantageux en raison de la structure assez simple
des fichiers. Pour les molécules inorganiques simples, les informations concernant la géométrie sont stockées
dans trois zones :
Si les molécules ne sont pas trop complexes, le calcul des coordonnées à partir des grandeurs expérimentales, ne nécessite que des connaissances
en géométrie élémentaire.
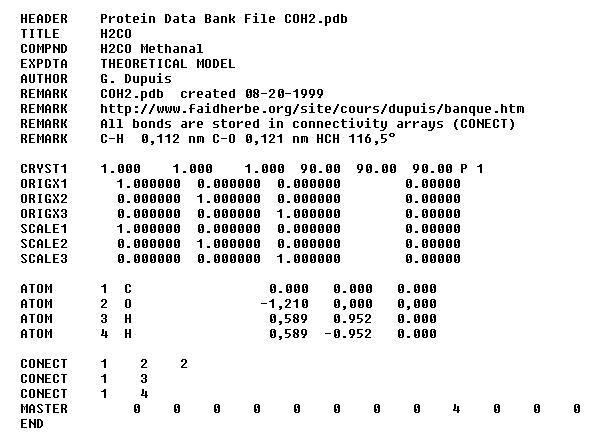
Voici un exemple très simple concernant la molécule de méthanal.
Cette zone contient le symbol de l'atome, le numéro qui lui est affecté et ses coordonnées cartésiennes
(x, y, z) dans un système d'axes orthonormé.
Dans cette zone sont stockées les informations concernant la connectivité. Une séquence telle que :
1 2 2 signifie que l'atome numéro 1 est doublement lié à l'atome numéro 2.
Dans cette ligne sont regroupées des informations servant au classement des fichiers. Les nombres qu'on y trouve représentent la somme
d'autres colonnes. Dans le fichier donné en exemple le chiffre 4 représente la somme du nombre des atomes de la molécule.
Vous pouvez, si vous le souhaitez, utiliser le contenu de cette page dans un but pédagogique et non commercial.
Gérard Dupuis - Lycée Faidherbe LILLE
octobre 2000