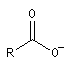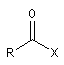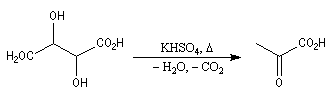|
|
|
|
|
|
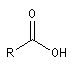
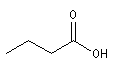
Acide |
Ion carboxylate |
Halogénure d'acyle |
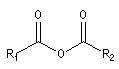
Anhydride d'acide |
|
|
|
|
|
|
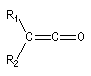
Cétène |
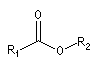
Ester |
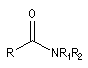
Amide |
Nitrile |
Cours de chimie Organique - G. Dupuis - Lycée Faidherbe de Lille
Acides carboxyliques
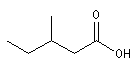
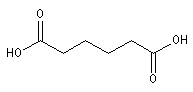
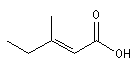
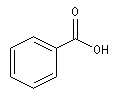
Nomenclature
|
|
|
|
|
|
Acide |
Ion carboxylate |
Halogénure d'acyle |
Anhydride d'acide |
|
|
|
|
|
|
Cétène |
Ester |
Amide |
Nitrile |
Même si le lien de parenté avec les acides carboxyliques est patent, leurs propriétés chimiques sont différentes de celles des acides, ce qui justifie une étude séparée.
|
|
|
|
Acide benzènecarboxylique |
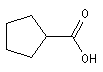
Acide cyclopentanecarboxylique |
Ions carboxylate Pentanoate d'ammonium Cyclohexanecarboxylate de sodium Etat naturel Acides gras
L'acide hexadécanoïque ou palmitique est un acide gras possédant 16 atomes de carbone. Le triester qu'il forme avec le glycérol est la palmitine.
Les noms des ions carboxylate sont formés à partir de ceux des acides parents en remplaçant le suffixe oïque par oate ou le suffixe ique par ate.
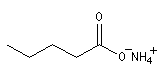
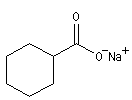
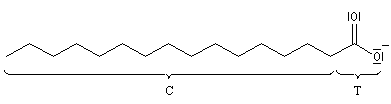
Les acides dont la molécule est constituée d'une chaîne de 4 à 36 atomes de carbone sont appelés acide gras. On peut les obtenir par saponification des graisses. Les esters des acides gras et du glycérol sont les triglycérides.

L'acide stéarique constitue un autre exemple d'acide gras qui entre dans la fabrication des bougies. On trouvera un résumé de ses principales propriétés à la référence [29].
|
|
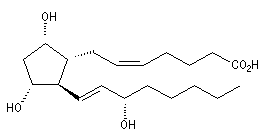
L'acide arachidonique est un acide gras insaturé possédant 20 atomes de carbone.
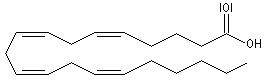 La conformation repliée de la chaîne met en évidence sa parenté entre avec la prostaglandine PGF2a dont il est un précurseur. |
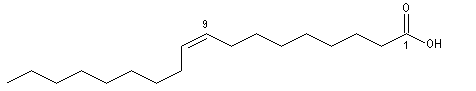
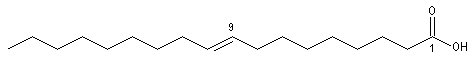
Les acides (Z)-octadéc-9-énoïque et (E)-octadéc-9-énoïque, sont des diastéréo-isomères. Leurs propriétés physico-chimiques sont différentes. On notera que la température de fusion de l'isomère E (élaïdique) est beaucoup plus grande que celle du Z (oléique) qui est liquide à la température ordinaire. L'acide oléique est l'acide gras le plus répandu à l'état naturel.
|
Formule |
Nom usuel |
TF/°C |
|
|
oléique |
13,4 |
|
|
élaïdique |
44 |
Les prostaglandines sont des acides gras insaturés dont le squelette possède 20 atomes de carbone. Elles ont un rôle d'hormones locales. La synthèse de la prostaglandine PGF2a représentée ci-dessous a été réalisée par plusieurs équipes. Les grandes étapes de la synthèse de G. Stork ont été vues dans le chapitre consacré aux composés conjugués.
Le degré d'insaturation des corps gras peut être déterminé par addition du réactif de Wijs sur les doubles liaisons suivie d'un dosage.
|
|
|
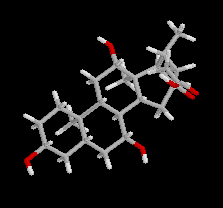
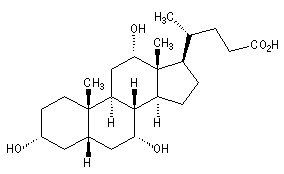
L'acide cholique a une structure voisine du cholestérol dont il dérive. Les groupes polaires -OH sont situés du même côté de la molécule ce qui permet de distinguer deux faces aux propriétés physico-chimiques différentes : la face regroupant les groupes polaires est hydrophile, l'autre est lipophile. En milieu biologique, il est combiné à un acide aminé comme le glycocolle dans la bile.
|
|
|
L'acide lactique est un hydroxyacide qui possède un intérêt industriel croissant. Il permet la synthèse d'un lactide dont la polymérisation conduit à un polymère biodégradable, appelé polylactide (PLA).
|
|
|
Acides importants sur le plan industriel
Acide acétique
La meilleure préparation industrielle de l'acide éthanoïque est la carbonylation du méthanol. Ce procédé, mis au point par Monsanto en 1971, utilise du trichlorure de rhodium comme
catalyseur [23].
![]()
Il permet la préparation de plus d'un million de tonnes d'acide éthanoïque par an. Il sert à préparer notamment l'acétate de vinyle.
Acide téréphtalique
L'acide téréphtalique est produit par oxydation du 1,4-méthylbenzène par l'oxygène de l'air en présence d'un catalyseur au cobalt.
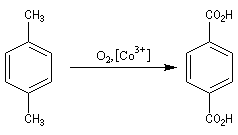
Propriétés physiques
Les températures d'ébullition des acides carboxyliques sont supérieures à celles des alcools de masse molaire comparable.
|
Alcool |
TE (°C) |
Acide |
TE (°C) |
|
CH3CH2OH |
78,5 |
HCO2H |
100,5 |
|
CH3CH2CH2OH |
97,2 |
CH3CO2H |
118 |
|
CH3CH2CH2CH2OH |
117,7 |
CH3CH2CO2H |
141 |
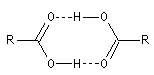
Une telle disposition géométrique, qui place les atomes d'hydrogène et d'oxygène dans le même alignement, rend ces liaisons hydrogène particulièrement solides.
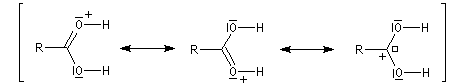
Structure des ions carboxylate acide méthanoïque d(C=O) = 0,123 nm d(C-OH) = 0,136 nm ion méthanoate d(C-Oa) = 0,127 nm d(C-Ob) = 0,127 nm Dans l'ion carboxylate, les longueurs des liaisons C-O sont égales et leur valeur est intermédiaire entre celle de la liaison C=O et de la liaison C-O dans
l'acide. Ce résultat peut être interprété en utilisant le modèle de la mésomérie. Les formes suivantes ont le même poids statistique ce qui témoigne d'une résonance importante : Savons
L'expérience montre que les atomes de carbone et d'oxygène d'un ion carboxylate sont dans un même plan. Pour l'acide méthanoïque et l'ion méthanoate, on relève les valeurs suivantes :
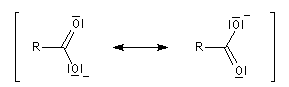
Les sels de sodium et de potassium des acides gras à longue chaîne carbonée en C12 et au delà, constituent les savons. On les obtient par la réaction de saponification
des graisses qui fournit en même temps le propane-1, 2, 3-triol ou glycérol.
Grâce à leur structure particulière constituée d'une longue chaîne carbonée hydrophobe et d'une tête polaire hydrophile, les savons sont capables d'émulsionner les
graisses en formant avec elles des micelles.
L'efficacité des savons est limitée en présence d'ions calcium du fait du caractère peu soluble des carboxylates de calcium qui précipitent. Une méthode de mesure de la dureté d'une eau consiste à mesurer l'épaisseur de la couche
de mousse qu'elle forme avec une solution aqueuse de savon de concentration connue.
![]()
Pour la même raison, on peut former facilement des précipités colorés en ajoutant des solutions ioniques de cuivre (II), de fer (II) ou de fer (III) à une solution aqueuse de savon.
Spectroscopie
Spectroscopie infrarouge Spectroscopie de RMN
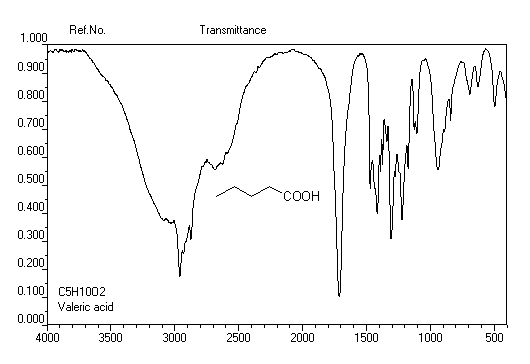
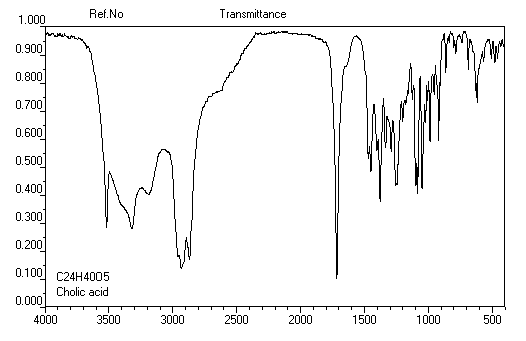
Du fait de la forte électronégativité du groupe carboxyle, le proton lié à ce groupe est très déblindé. Le déplacement chimique
apparaît souvent au delà de d = 11 ppm. Du fait de la mobilité protonique et du phénomène d'échange avec le solvant,
la position du pic correspondant dépend des conditions expérimentales.
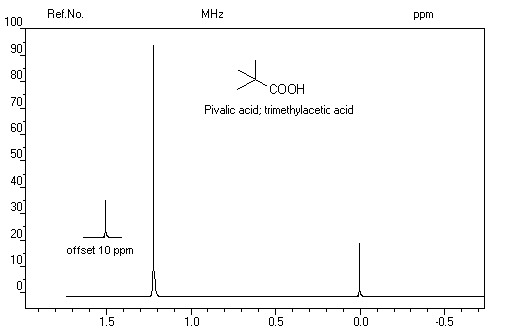
Comme dans le cas des alcools, l'addition d'eau lourde fait intervenir l'équilibre :
Les protons portés par l'atome de carbone en a du groupe carboxyle sont déblindés vers d = 3,5 ppm en raison du caractère électronégatif de ce groupe.
Réactions de Piria et de Ruzicka La réaction est possible avec des diacides. On a alors à la fois déshydratation et décarboxylation. La formation de cycles à 5 ou 6 chainons est favorisée comme l'illustre l'exemple suivant extrait de la synthèse historique du terpinéol (W. H. Perkin 1904). Réaction de Hunsdiecker La réaction de Hunsdiecker procède vraisemblablement d'un mécanisme radicalaire : Voici un exemple d'application de la réaction de Hunsdiecker [28] : Une réaction très comparable consiste à partir de l'acide et à utiliser le dibrome en présence d'oxyde de mercure [30].
La décarboxylation des sels de calcium des acides carboxyliques est réalisable à température élevée. Cette réaction a été découverte par le chimiste Italien R. Piria.
Elle fournit des cétones. Ainsi, la propanone était préparée autrefois par pyrolyse de l'acétate de calcium ce qui explique son nom courant : l'acétone.
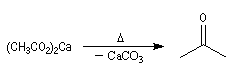
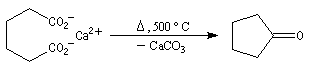
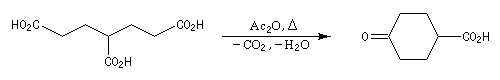
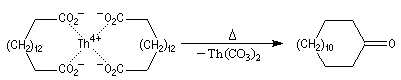
La décarboxylation des carboxylates d'argent en présence de dibrome, constitue une méthode de passage des acides carboxyliques aux dérivés bromés.
La découverte de cette réaction est attribuée au chimiste et musicien russe A. Borodine en 1861. Elle est plus connue sous le nom de réaction de Hunsdiecker. Sa force motrice est la formation
de composés stables CO2 et AgBr.
![]()
![]()
![]()
![]()
![]()
![]()
Décarboxylation des b-cétoacides Décarboxylation des a-cétoacides Le bilan de ces réactions s'écrit :
La décarboxylation des b-cétoacides s'opère dans des conditions de température modérées entre 50 °C et 60 °C. La formation d'une liaison
hydrogène intramoléculaire permet la création d'un état de transition cyclique.
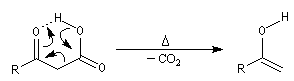
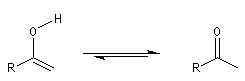
En milieu biologique, la décarboxylation de l'acide pyruvique en éthanal et CO2 est une étape de la fermentation alcoolique. Elle s'effectue avec le concours d'une enzyme : la pyruvate décarboxylase.
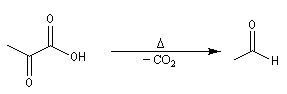
![]()
![]()
![]()
![]()
![]()
Elimination-décarboxylation Propriétés acido-basiques Acidité Ka
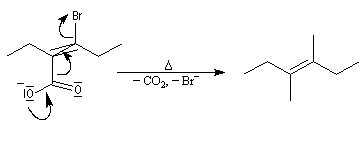
Les ions carboxylate des acides b-halogénés sont facilement décarboxylés par chauffage. L'ion carboxylate et l'atome de brome doivent
être dans une position anticoplanaire pour que l'élimination se produise.
Les acides carboxyliques sont des acides au sens de Brönsted. Ils réagissent avec l'eau pour donner des ions carboxylate et des ions oxonium.
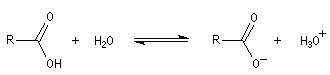
|
Acide |
HCO2H |
CH3CO2H |
C2H5CO2H |
C3H7CO2H |
|
pKa |
3,75 |
4,75 |
4,88 |
4,82 |
Lorsqu'on étudie les pKa des différents acides chloroéthanoïques, on observe une décroissance du pKa du couple avec l'augmentation du nombre d'atomes de chlore. Les résultats expérimentaux sont donnés ci-dessous à 298 K :
|
Acide |
CH3CO2H |
ClCH2CO2H |
Cl2CHCO2H |
Cl3CCO2H |
|
pKa |
4,75 |
2,87 |
1,20 |
0,52 |
Il est naturellement tentant de mettre en relation la force des acides avec la fragilisation de la liaison O-H dûe à l'effet inductif -I exercé par les atomes de chlore. Cela a souvent été écrit dans le passé. Il importe de bien distinguer la situation en phase gazeuse et celle qu'on rencontre dans un solvant comme l'eau où la solvatation des espèces chargées joue un rôle essentiel.
Basicité
L'étude par cryoscopie montre que dans l'acide sulfurique concentré, l'acide éthanoïque est complètement protoné. Compte-tenu du pKa voisin de -6 pour le couple acide base correspondant, la protonation ne peut avoir lieu qu'en présence d'acides concentrés nivelés par l'eau dont les couples ont un
pKa < 0.
Remarque : les acides plus forts que l'acide sulfurique concentré sont appelés superacides.
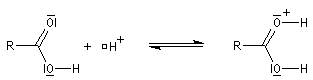
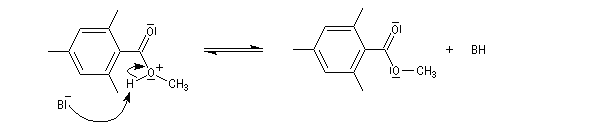
Chez les acides présentant un encombrement stérique très important comme l'acide mésitoïque, la protonation s'effectue sur le groupe -OH. Ce phénomène joue un rôle important dans l'estérification de ce type d'acide par un mécanisme inhabituel (AAC1).
Propriétés nucléophiles des ions carboxylate
Acylation des ions carboxylate Alkylation par un dérivé halogéné L'exemple suivant constitue un exemple de réaction utilisant une catalyse par transfert de phase.
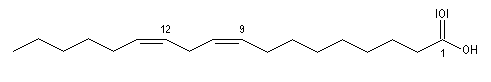
Méthylation par le diazométhane Une réaction du même type permet la méthylation des alcools. En revanche la réaction entre le diazométhane et un chlorure d'acyle fournit une a-diazocétone. Le mécanisme de cette réaction est détaillé dans le chapitre particulier consacré au composés éthyléniques. Le cas de l'acide linoléique est un peu plus compliqué.
Les ions carboxylate sont de bien meilleurs nucléophiles que les acides carboxyliques. La réaction d'un ion carboxylate sur un chlorure d'acyle permet de préparer des anhydrides symétriques ou mixtes.
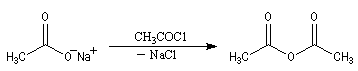
On peut aussi alkyler les ions carboxylate en utilisant un halogénure d'alkyle primaire ou secondaire. On prépare ainsi des esters. Avec un halogénure tertiaire on observe essentiellement une réaction d'élimination.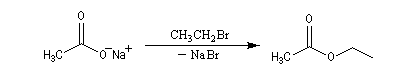
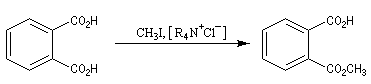
Une méthode particulièrement douce pour méthyler les ions carboxylate consiste à les faire réagir avec le diazométhane. Le seul sous-produit de la réaction est le diazote qui s'échappe du milieu réactionnel. Cette réaction nécessite des précautions particulières sur le plan expérimental car le diazométhane est un agent méthylant extrêmement toxique.
![]()
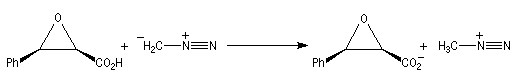
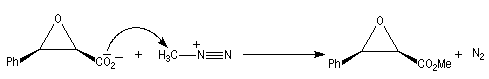
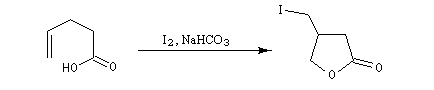
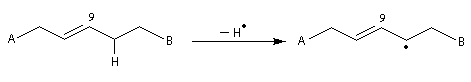
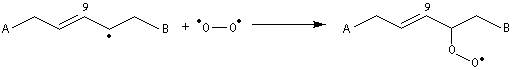

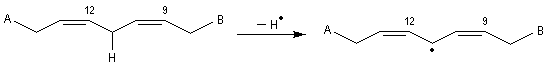
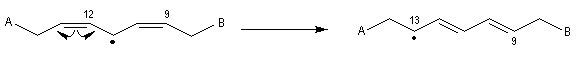
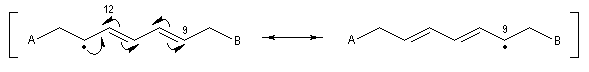
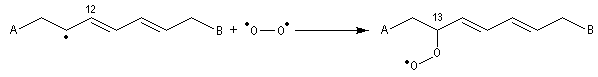
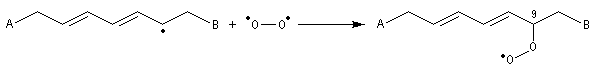
Propriétés nucléophiles des acides
Déshydratation des acides
La déshydratation des acides en présence d'un déshydratant énergique comme P4O10 conduit aux anhydrides.
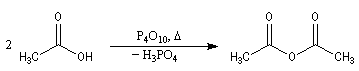
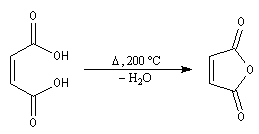
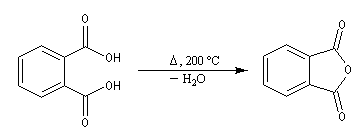
![]()
Passage aux dérivés d'acides On trouvera une méthode de préparation du chlorure d'acyle de l'acide oléique à la référence [32]. Réduction des acides Réduction par H2 Réduction par LiAlH4 Le bilan de ces réaction est écrit ci dessous :
La réaction peut être effectuée en utilisant différents agents halogénants. Ces sont les mêmes que ceux qu'on utilise pour transformer les
alcools en dérivés halogénés :
![]()
![]()
![]()
![]()
La réduction du groupe carboxyle par le dihydrogène n'est réalisable que dans des conditions sévères de température et de pression en présence de catalyseurs
tels que Ni, Pt ou Ru. On peut donc effectuer l'hydrogénation catalytique sélective d'une double liaison éthylénique en présence d'un groupe carboxyle en choisissant convenablement les conditions
expérimentales.
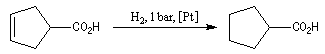
LiAlH4 réduit les acides en alcools.
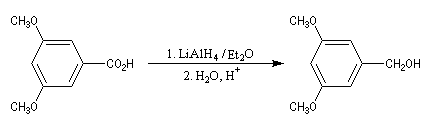
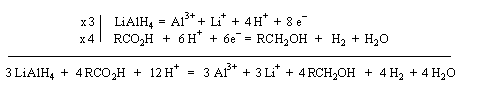
Il est souvent préférable d'estérifier la fonction acide et réduire ensuite l'ester obtenu.
Réduction par le couple BH3, Me2S
Les acides peuvent être réduits en alcools par le couple diborane-diméthylsulfure BH3, Me2S et BF3/Et2. L'un des avantages de cette réaction réside dans la chimiosélectivité du réactif. Une condition importante pour le succès de la
réaction est l'absence de double liaison ou de triple liaison carbone-carbone. Celles-ci réagissent en effet avec BH3 pour donner des organoboranes.
Dans l'exemple suivant la fonction acide est réduite en alcool. La fonction ester est inaltérée.
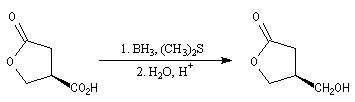
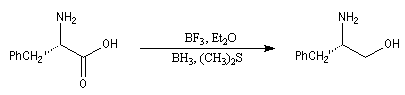
La réaction du S-phénylalanol avec l'anhydride acétique permet la synthèse de la (S)-4-phénylméthyl-2-oxazolidinone. L'alkylation des oxazolidinones permet la préparation de composés énantiopurs par la méthode d'alkylation énantiosélective d'Evans.
Additions nucléophiles sur le carbonyle
Composés organolithiens
L'une des réactions les plus importantes des acides carboxyliques avec les composés organométalliques, est
celle avec les organolithiens qui conduit aux cétones. Dans un premier temps, le lithien réagit comme une base en arrachant l'atome d'hydrogène du groupe carboxyle. On obtient un carboxylate de lithium.
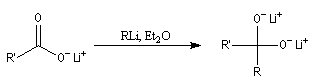
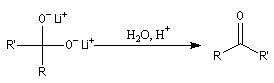
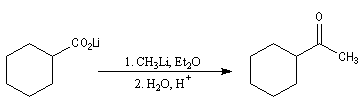
Réactions avec les alcools primaires et secondaires
La réaction entre un acide carboxylique et un alcool est appelée estérification de Fischer. Il s'agit d'une réaction lente et limitée nécessitant
l'utilisation d'un catalyseur. L'équilibre peut être déplacé en utilisant un appareil de Dean-Stark. Le mécanisme AAC2 de cette réaction a été vu dans le chapitre consacré aux alcools.
Formation de lactones
Avec un composé bifonctionnel comportant une fonction acide et une fonction alcool, l'estérification peut avoir lieu de façon intramoléculaire. L'ester cyclique obtenu est appelé lactone. L'équilibre de lactonisation est favorable pour les cycles possédant 5 ou 6 chaînons et les hydroxy-acides correspondants se cyclisent spontanément.
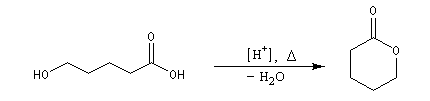
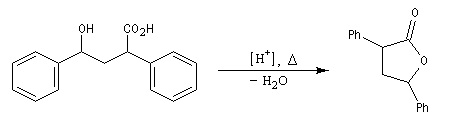
La synthèse de composés bicycliques est possible dans les cas favorables comme dans cette étape de la synthèse du terpinéol.
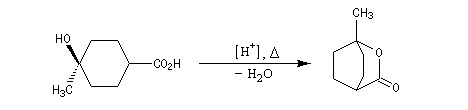
La réaction peut impliquer la forme énolique d'un céto-acide comme dans le cas de l'angelica lactone qui peut être obtenue par cyclisation de l'acide 4-oxopentanoïque (acide lévulique).
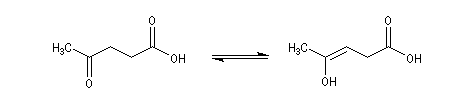
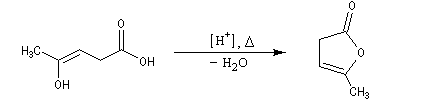
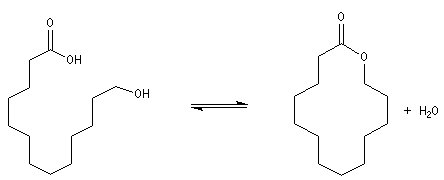
Davantage d'informations sur la synthèse des grands cycles par lactonisation : [33]
La réduction des lactones fournit des hémiacétals
Formation de lactide
Dans l'acide lactique les fonctions acide et alcool sont trop proches pour qu'une cyclisation en lactone puisse avoir lieu. En revanche, deux molécules d'acide lactique peuvent réagir entre-elles. On obtient un composé cyclique appelé lactide.
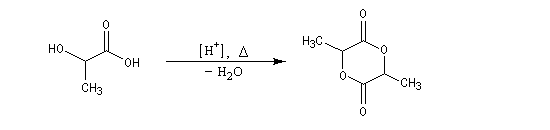
La polymérisation de ce composé conduit à un polymère biodégradable appelé PLA, [31].
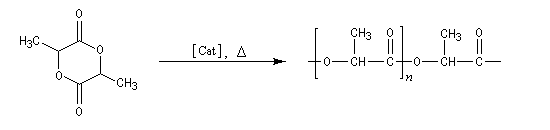
|
|
L'étude par cryoscopie de solutions d'acide 2, 4, 6-triméthylbenzoïque (mésitoïque) dans l'acide sulfurique concentré, montre un abaissement du point de
congélation double de celui qu'on obtient avec l'acide acétique. On en déduit qu'il s'est formé 4 particules dans la solution. |
Le mécanisme est le suivant :
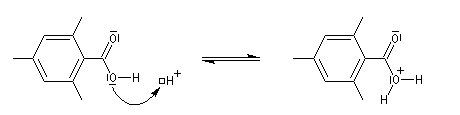
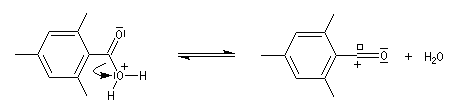
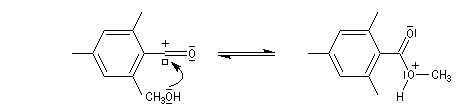
Ammoniac et amines non tertiaires
La réaction entre un acide carboxylique et une amine ou l'ammoniac conduit à la formation d'un carboxylate d'ammonium ou d'aminium au terme d'une réaction acido-basique.
![]()
![]()
Les plupart des amides acycliques sont préparés par acylation des amines ou de l'ammoniac en utilisant des dérivés d'acides.
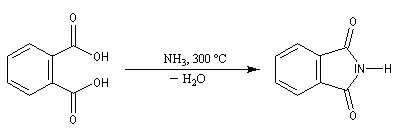
Réactions au voisinage du carbonyle
Réaction de Hell-Vohlardt-Zelinsky La réaction s'effectue en plusieurs étapes : Les acides a-halogénés comme substrats Bibliographie
Les acides peuvent être bromés en présence de dibrome et de phosphore rouge.
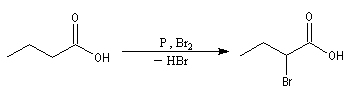
![]()
![]()
![]()
Les acides a-halogénés sont largement utilisés en tant que composés intermédiaire en synthèse organique. Ils constituent
en effet des substrats de choix pour réaliser des substitutions nucléophiles, du fait de la grande mobilité de l'halogène en a du groupe carboxyle. Voici quelques exemples :
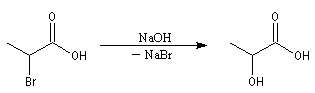
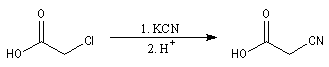
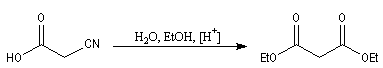
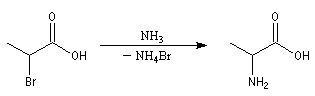
[2] P. Rendle, M. V. Vokins, P. Davis, Experimental Chemistry (Edward Arnold).
[3] L. F. Fieser, K. L. Williamson - Organic Experiments (D. C. Heath and Company).
[4] R. Adams, J. R. Johnson, C. F. Wilcox - Laboratory Experiments in Organic Chemistry (The Macmillan Company, Collier-Macmillan Limited).
[5] G. K. Helmkamp, H. W. Johnson Jr, Selected Experiments in Organic Chemistry (W. H. Freeman and Co).
[6] Vogel's Textbook of Practical Organic Chemistry (Longman).
[7] J. R. Mohrig, D. C Neckers, Laboratory Experiments in Organic Chemistry (D. Van Nostrand Company).
[8] R. Q. Brewster, C. A. Van der Werf, W. E. Mc Ewen, Unitized Experiments in Organic Chemistry (D. Van Nostrand Company).
[9] F. G. Mann, B. C. Saunders, Practical Organic Chemistry (Longman).
[10] M. T. Yip, D. R. Dalton, Organic Chemistry in the Laboratory (D. Van Nostrand Company).
[11] Miller, Neuzil, Modern Experimental Organic Chemistry (DC Heath, 1982)
[13] A. Streitwieser Jr, C. H. Heathcock - Introduction to Organic Chemistry, Macmillan Publishing Co.
[14] J. March - Advanced organic chemistry, Wiley Interscience.
[15] F. A. Carey, R. S. Sunberg - Advanced Organic Chemistry, Plenum Press 1990.
[17] N. T. Ahn - Introduction à la chimie moléculaire, Ellipses 1994.
[18] R. Brückner - Mécanismes réactionnels en chimie organique, De Boeck Université 1999.
[20] P. Laszlo - Logique de la synthèse organique. Cours de l'Ecole Polytechnique, Ellipses, 1993.
[21]Peroxyl Radicals, Z B Alfassi, ed., John Wiley & Sons, New York, 1997.
[23] The Monsanto Acetic Acid Process
[24] L. Ruzicka the Nobel Prize in Chemistry 1939
[25] Nomenclature des acides carboxyliques
[26] S-4-(phenylmethyl)-2-oxazolidinone by J. R. Gage and D. A. Evans
[27] cyclohexylméthylcétone by T. M. Bare and H. O. House
[28] Hunsdiecker reaction by C. F. H. Allen and C. V. Wilson
[29] Stearic acid and sodium stearate by H. Rzepa
[30] Bromocyclopropan by John S. Meek and David T. Osuga
[31] Synthèse du PLA
[32] Oleoyl Chloride by C. F. H. Allen, J. R. Byers, Jr., and W. J. Humphlett
[33] Macrolactonization by Thomas Boddaert.
[34] Physiopathologie du stress oxydant par J. Cillard, Faculté de Pharmacie, Université de Rennes.
[35] Peroxydation des lipides.
Vous pouvez, si vous le souhaitez, utiliser le contenu de cette page dans un but pédagogique et non commercial.
Texte, dessins, photographies : Gérard Dupuis - Lycée Faidherbe de LILLE
avril 2014